錒系元素的非尋常氧化態化學Ⅰ.氣相、固相和水溶液
2020-12-30 09:48:04蘇一茂黃聞亮
核化學與放射化學 2020年6期
鄧 翀,蘇一茂,黃聞亮
北京大學 化學與分子工程學院,北京分子科學國家研究中心,稀土材料化學及應用國家重點實驗室, 放射化學與輻射化學重點學科實驗室,北京 100871
錒系元素是89號元素錒到103號元素鐒共15個金屬元素的總稱。傳統上,前七個元素(錒至镅)統稱為前錒系元素,而后八個元素(鋦至鐒)統稱為后錒系元素。錒系元素約占目前元素周期表中元素總數的1/8,并與鑭系元素合稱f區元素。在錒系元素中,釷和鈾在地殼中有一定的分布[1];此外,錒和鏷在自然界中也有極少的存量。其他錒系元素最早都是通過人工核反應發現的,它們的合成往往涉及連續中子俘獲與β衰變,或者輕核(4He、11B、12C、15N、16O等)轟擊過程。局限空間內的高能核爆也可以產生超鈾元素,锿和鐨最早就是在氫彈爆炸后的輻射落塵中被發現的[2]。所有錒系元素的同位素均具有放射性[3]。這使得它們有著豐富的核反應性,尤其是自發或誘導的核裂變性質。這一方面促成了以鈾為主的核能利用,但另一方面也使錒系元素的化學研究必須在一定的輻射防護條件下進行。因此,錒系元素的化學研究,相比于擁有穩定同位素的元素,更為受限。232Th和238U在錒系核素中有著最高的穩定性,其α衰變的半衰期大于109a;同時,它們也是天然釷和鈾中占比最高的同位素,因此二者被廣泛地用于化學研究[3]。
當前,常規價態的錒系元素配位化學已經不足以滿足錒系化學發展的需求。對于非尋常價態的錒系化學的追求不僅可以滿足化學家打破邊界的好奇心,同時還有著廣泛的應用前景。比如,實驗上發現將Bk3+氧化至Bk4+有利于锫的分離[4]。本綜述(第一部分)將從氣相、固相和水溶液等方面勾勒錒系元素非尋常氧化態化學的發展歷史與最新進展,首先分析錒系元素電子構型的特點,并給出文中所涉“非尋常氧化態”的定義與范圍。對于研究較多的元素,如釷、鈾等,將單獨進行介紹,而其他錒系元素則分為前錒系與后錒系兩類分別介紹。
1 錒系元素的電子構型與氧化態
錒系金屬原子及其三價和四價陽離子的基態價電子構型列入表1。對于錒、釷原子,由于6d軌道能量低于5f軌道,電子優先填充在6d軌道上。從鏷開始,6d軌道上只占據一個電子,5f軌道開始填占電子。從钚開始,除了5f76d17s2構型的鋦和5f146d17s2構型的鐒以外,6d軌道不再填占電子,新增的價電子均填入5f軌道。錒系金屬原子對應的三價和四價陽離子則具有[Rn]5fn基態電子構型[3]。不同于稀土金屬收縮于離子核內部的4f軌道,錒系金屬的5f軌道基本不被6s和6p亞層所屏蔽,而5f軌道比4f軌道在原子核外也更加擴展[5]。2000年以來,對相似體系中鑭系/錒系金屬配合物的對比研究,也證實了5f軌道較4f軌道能更好地參與形成共價鍵[6-8]。
對于前錒系元素,7s、7p、6d、5f軌道的能量差異不大,這些幾乎簡并的軌道都有可能參與到化學反應過程中。此外,錒系金屬的5fn7s2和5fn-16d17s2電子構型之間的能量差顯著小于鑭系金屬,使得錒系金屬還可能以不同的電子構型參與反應。這些特點使得前錒系元素可以在化合物中表現出豐富的氧化態[9]。對于鈾及其之前的前錒系元素,最穩定的氧化態是各自的最高氧化態(即失去全部價電子),分別為:Ac(Ⅲ)、Th(Ⅳ)、Pa(Ⅴ)、U(Ⅵ)。對于镎、钚、镅而言,最穩定的氧化態分別為+5、+4、+3[10]。由于5f電子之間并不能有效地相互屏蔽核電荷,因而隨著錒系元素的原子序數增大,5f電子與核結合得更加緊密,其能量急劇降低。這一方面使得基態原子的價電子優先填入5f軌道[9],另一方面也使得5f電子在化學反應中更難失去。因此,镎、钚、镅等無法以+7或更高氧化態穩定存在。5f能量驟降帶來的影響更突出地體現在后錒系金屬離子的氧化還原性質上。除了少數幾個元素可以在相對溫和的條件下實現變價(如鍆、锘的介穩/穩定二價和锫的介穩四價),后錒系元素的化學行為在大部分情況下均由正三價主導。這一點類似于鑭系元素,使得后錒系元素與鑭系元素在化學性質上非常相近,給核燃料后處理過程中二者的分離造成了困難[11-12]。
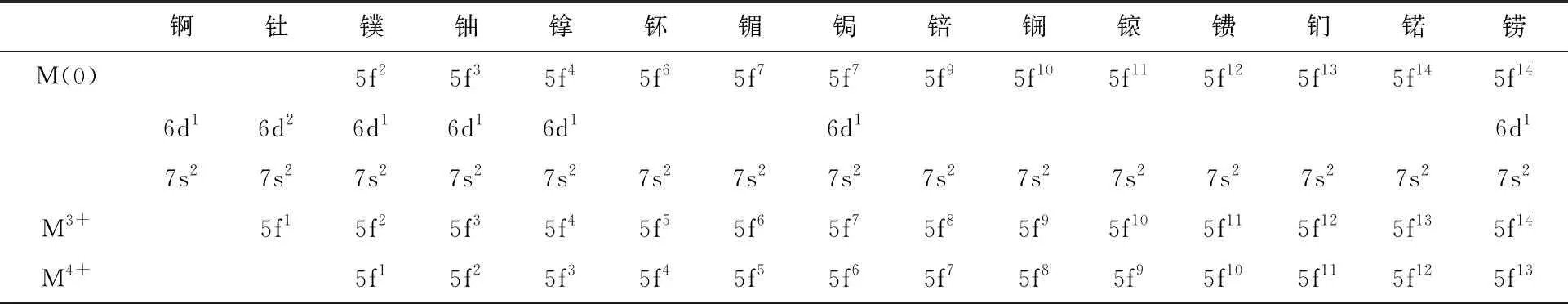
表1 錒系金屬原子及三價和四價離子的基態價電子構型Table 1 Ground state electron configuration of M(0), M3+ and M4+ of actinides
三價錒系金屬的六配位有效離子半徑(r(M3+))[13]及其M3+/2+和M4+/3+電對的標準還原電勢(E)[14]列入表2。借鑒于稀土金屬非傳統氧化態的歸類,原則上應當將比二價釤更難還原得到(E(Sm3+/2+)=-1.55 V vs.一般氫電極(NHE))或比四價鈰更難氧化得到(E(Ce4+/3+)=+1.7 V vs. NHE)[16]的錒系金屬的氧化態作為“非尋常氧化態”。不過由于錒系金屬的氧化態變化遠比稀土金屬復雜,并且其氧化還原電勢顯著受到環境(溶液酸堿性、配體體系等)影響,因此接下來將展開進行具體討論。低價方面,E(M3+/2+)從錒到锘遞升,其變化范圍明顯大于鑭系金屬系列[17]。考慮到絕大部分錒系金屬的E(M3+/2+)具有相當大的負值絕對值,將錒系金屬的二價認定為“非尋常氧化態”,也包括較易得到的Md(Ⅱ)、No(Ⅱ)以作對比。此外,后錒系元素的E(M4+/3+)均接近或遠高于E(Ce4+/3+),因此將從鋦開始的后錒系金屬的四價及更高氧化態均歸為“非尋常氧化態”。考慮到四價釷和鏷很難被還原至三價(E(Th4+/3+)=-3.8 V,E(Pa4+/3+)=-2.0 V vs. NHE),將釷、鏷的三價也納入“非尋常氧化態”。
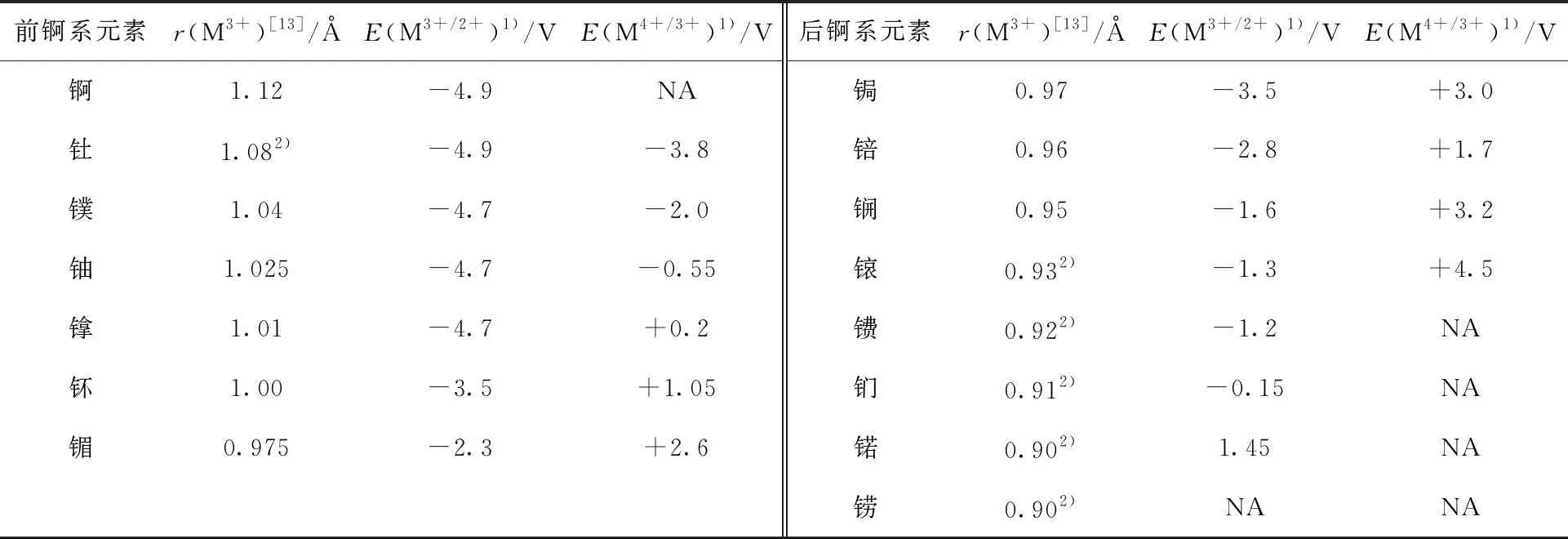
表2 三價錒系金屬離子半徑與M3+/2+、M4+/3+還原電勢(vs. NHE)[14]Table 2 Ionic radii of M3+ and standard reduction potentials for M3+/2+and M4+/3+ (vs. NHE)[14]
綜上分析,將Ac(Ⅱ)、Th(Ⅱ)/Th(Ⅲ)、Pa(Ⅱ)/Pa(Ⅲ)、U(Ⅱ)、Np(Ⅱ)、Pu(Ⅱ)、Am(Ⅶ)、M(Ⅱ)/M(Ⅳ)(M=Cm、Bk、Cf、Es、Fm、Md、No、Lr)等視為錒系元素的“非尋常氧化態”,并在本綜述中予以討論。其中包括已經得到結構或光譜等表征確認的氧化態(如Th(Ⅲ)、U(Ⅱ)),也包括當前對其存在尚有爭論的氧化態(如Pu(Ⅷ)、Md(Ⅰ))。
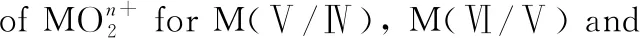
表3 錒酰離子相關的M(Ⅴ/Ⅳ)、M(Ⅵ/Ⅴ)、 M(Ⅶ/Ⅵ)還原電勢(vs. NHE)1)[14]
2 氣相
2.1 釷
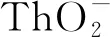
2.2 鈾
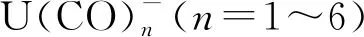
2.3 其他前錒系元素
(1) 鏷
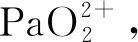
(2) 钚
Green等[47]利用濺射設備在低溫稀有氣體基質中分離了PuO等钚氧化物,并測定了紅外光譜。Domanov等[48]利用氣相熱色譜技術研究了微量钚與氧氣在氦載氣中的反應,得到的易揮發物種被指認為PuO4。雖然PuO4和PuF8、PuO2F4均被認為是可能的Pu(Ⅷ)目標分子[49],但表征數據的缺失使得PuO4存在的真實性存疑。Zaitsevskii等[50]的計算研究表明,PuO4的穩定性高于AmO4,但不及雙金屬化合物PuAmO7。利用稀有氣體基質的穩定化作用有可能在4~6 K得到PuO4、AmO4[51],而Gong等[52]關于IrO4的氣相合成也為此提供了技術參考。關于PuO4的真實電子結構,Huang等[53]指出,盡管钚和釕、鋨一樣具有八個價電子,但PuO4并不存在與RuⅧO4、OsⅧO4相似的穩定電子構型。通過多種量子化學計算方法,他們發現PuO4的基態是C2v對稱的(PuⅤO2)+(O2)-五重態,而D4h對稱的PuⅧO4只是亞穩態。類似的“降價”現象同樣出現在PuOnF8-2n(n=0~4)體系中,但這一體系中不同電子結構的能量差值更小[54]。
(3) 镅
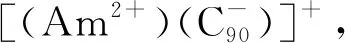
2.4 后錒系元素
(1) 鋦
Smith等[56]最早發現Cm2O3在高溫下的氣相解離產物主要是CmO。CmO的等電子體,CmF+,也曾在Cm+與六氟丙烯的氣相反應中被發現[57]。通過引入硝酸根配位,Kovcs等[58]首次在氣相中穩定了五價鋦酰離子:[CmO2(NO3)2]-。在氣相能與水分子反應,首先形成的[CmO2(H2O)4]+經氧交換過程轉化為高價鋦氟化物的氣相化學十分復雜,雖然早期的熱色譜研究發現了CmF6、CmOF3和EsF4存在的證據,但其中錒系金屬的氧化態卻無法清楚指認[60]。Domanov等[61]利用氣相熱色譜技術,研究了微量CmHx(2≤x≤3)在氦載氣中與O2的反應,得到的易揮發鋦化合物因與PuO4有某些相似的性質而被指認為CmⅧO4。但是,由于缺少對金屬氧化態的直接表征,這一結論尚存在爭議[51]。
(2) 锫、锎、锿與鐨、鍆、锘、鐒
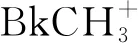
3 固相
3.1 釷
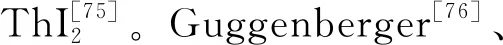
釷的低價氧化物尚無在固相存在的可靠證據,但釷與硫、硒、碲可以形成多種低價化合物[78]。以硫為例,真空中高溫加熱ThS2可以獲得Th7S12;ThS2與釷氫化物在還原氣氛下高溫加熱可得到ThS;在釷氫化物和H2S的反應產物中發現了Th2S3[79]。在加熱條件下,釷粉或釷屑還可與氮氣或氨氣反應生成ThN[80],其薄膜具有典型的金屬性[81]。釷單質在高溫下可與Th3P4反應生成ThP[82],而ThAs和ThSb則可由單質直接反應得到[78]。
3.2 鈾
2018年Havela等[83]利用反應濺射沉積的方法合成了UH2薄膜,并發現其是一種金屬性的鐵磁體。X射線光電子能譜(XPS)研究表明,UH2和UH3的電子結構有一定的相似性。低價鈾的氧化物(如UO)早在1950年就被發現[84],其中UO相需要與UC、UN等共存才能穩定。但是,到目前為止UO在固相的結構尚不明確[85]。Eastman等[79]發現,通過將US2與鈾氫化物混合加熱至2 000 ℃以上,或利用鈾氫化物與H2S在一定條件下反應,均可以制備US。此外,Kulyukhin等[86]還發現,在氯化物熔鹽中,Nd2+可以還原U3+。Langridge等[87]向USb中引入Te,發現得到的物質中有UTe相,并通過磁學研究推測有U2+存在。U2+在CaF2等氟化物晶體中的摻雜[88-89],以及U2+與Gd2Cl3共結晶的研究也有相關報道[86]。但是,目前尚沒有充分的證據可以指認這些固相化合物中鈾的氧化態。
3.3 其他前錒系元素
(1) 錒
盡管1970—1980年代的系列研究揭示了絕大部分f區元素都能以+2氧化態存在于氯化物熔鹽中,但二價錒目前尚無在熔鹽中形成的證據[90]。
(2) 鏷
金屬鏷在氫氣中250 ℃加熱可以得到PaH3,而調節反應溫度和氫氣的壓力可得到組成為PaHx(x=1.3~3或2~3)的相或Pa3Hx(x=4~5)的固溶體[91]。PaI3是目前唯一報道的低價鏷鹵化物,在300 ℃下加熱PaI5即可得到。它具有和CeI3相似的粉末X射線衍射(PXRD)圖譜[92]。低價鏷的氧化物還沒有獨立報道,但Zachariasen等[93]曾發現金屬鋇還原PaF4的副產物之一是PaO,并發現其具有NaCl型結構。此外,利用PaI5熱解離反應制備的新鮮金屬鏷,能夠與氮氣在1 100 ℃反應得到PaN[94];而從鏷與砷的單質出發,經過碘的化學氣相輸運反應可以得到PaAs[95]。
(3) 镎、钚
金屬镎能與氫氣在不同溫度、壓力條件下反應。隨著氫氣含量的增加,產物逐漸由NpH2+x(x=0~0.7)轉變為NpH3[96-97]。金屬钚與氫氣的反應則生成組成連續的固溶體,并不存在PuH2、PuH3的純相[98]。磁學研究、XPS/俄歇電子能譜(AES)、中子粉末衍射等結果表明,形式上的低價或混價钚氫化物中钚仍以三價形態存在,其準確的電子結構可描述為Pu3+(H-)x(e-)3-x(x<3)[99-101]。Mikheev等[102]曾報道在SrCl2熔鹽中用PrCl2還原PuCl3,得到了PuCl2,并推測出Pu3+/Pu2+電對的還原電勢為-2.59 V(vs. NHE)。但后來關于Pu(Ⅲ)、Np(Ⅲ)在熔鹽中的電化學還原研究均不支持二價中間物種的形成[103-105]。將Np2S3與金屬镎混合后高溫加熱[106],或者進行镎和硫單質的蒸氣反應,均可以得到具有NaCl型結構的NpS[107]。此外,PuS則可以通過鈣蒸氣還原PuF3或钚粉與H2S反應制得[98]。
(4) 镅
在加熱條件下,金屬镅與氫氣反應生成AmH3和AmH2+x兩種氫化物,后者與NpH2+x和PuH2+x同構[108]。XPS研究確認了AmH2+x中二價镅的存在[109]。Edelstein等[110]曾將Am2+摻雜到CaF2晶體中,并對其進行了電子順磁共振(EPR)與可見吸收光譜的表征。Baybarz等[111-112]隨后利用金屬镅與鹵化汞的置換反應,合成了AmCl2、AmBr2與AmI2,并通過磁學測量確認了其中存在Am2+。镅的低價氧化物AmO有可能通過金屬镅和Am2O3的歸中反應得到,而AmH3與硫單質、硒單質或碲化物共熱,則可以得到AmS、AmSe或AmTe[20]。理論計算表明,在這些硫族元素化合物中,僅AmTe中存在Am2+,而且其中镅的半充滿5f7電子構型的能量也僅比5f66d1電子構型(其中6d電子離域到導帶中)的能量低0.1 eV[113-114]。在熔鹽體系中,有較多關于二價镅的研究:比如,金屬镅在NaCl-KCl熔鹽中與PuCl3反應可以得到AmCl2[115];Am(Ⅲ)在LiCl-KCl、NaCl-2CsCl等熔鹽中的電化學還原反應會經歷Am(Ⅱ)的形成[116-117]。Am(Ⅱ)在熔鹽中的易得性與穩定性,已經在镅與鋦和鑭系金屬的新型分離技術中得到了利用[118]。
3.4 后錒系元素
(1) 鋦
1970年,Bansal等[119]通過在純氫氣中加熱金屬244Cm,得到了首例鋦的氫化物。通過比較其與MH2+x(M=Np、Pu、Am)的XRD圖譜,他們認為產物是CmH2+x(c≤x≤0.7,晶胞參數c以nm為單位)。Gibson等[120]后來利用鋦的更穩定核素248Cm制備了兩種晶胞參數不同的氫化物,248CmH3-δ和248CmH2+x,并通過與鑭系金屬氫化物體系的對比確認了鋦的二氫化物的存在。Damien等[121]還發現,金屬鋦與硫族元素單質在高溫下反應下能形成CmA(A=S、Se、Te)等二元硫族元素化合物,但無法得到純相,而在較低溫度下加熱,得到的產物是三價鋦的化合物。
四價鋦化合物都是以氧化物或氟化物形式存在[122]。1955年,Asprey等[123]利用400 ℃下CmF3、Cm2(C2O4)3在氧氣中的燃燒反應制備了244CmO2。Morss等[124]對248CmO2進行了中子衍射研究,結果表明化合物的組成為CmO1.99±0.01,但其有效磁矩(3.36±0.06)μB卻符合Cm(Ⅲ)的特征。后來的計算研究表明,在CmO2中,鋦的氧化態處于+4(f6)和+3(f7)之間,其f電子相比前錒系金屬的二氧化物有著更高的離域特征[125]。
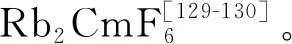
(2) 锫
1972年,Fahey等[133]在石英毛細管中加熱金屬锫和氫氣,得到了組成為BkH2+x(x<1)的锫的氫化物,屬立方晶系。Gibson等[134]進一步深入研究了金屬锫和氫氣的反應體系,發現在600 ℃加熱條件下生成BkH2+x,而在300 ℃下則生成六方晶系的BkH3-δ。他們據此確認了Bk(Ⅱ)的氫化物BkH2+x的存在,并認為x取值可能在-0.2~+0.3。
固相中四價锫的穩定性較高,BkO2、BkF4、Cs2BkCl6在1980年代前已得到報道[135]。BkO2具有CaF2型面心立方結構,其價帶XPS譜主要體現為5f電子發射的特征[136]。Asprey等[130]利用氟氣作氧化劑制備了BkF4。Morss等[137]發現,將Bk(OH)4溶解在濃鹽酸后與CsCl反應可以得到Cs2BkCl6,其與Rb2MnF6具有相似結構;而四甲基氯化銨可以替代CsCl作為沉淀劑,得到的[N(CH3)4]2BkCl6具有更低的溶解度和更高的穩定性[138]。此外,盡管在早期的Bk(Ⅳ)共沉淀實驗中,經常引入碘酸鹽作為沉淀劑[139-140],但BkⅣ(IO3)4的合成直到2017年才由Albrecht-Schmitt等實現。他們發現,向磚紅色的Bk(OH)4中加入碘酸,最終可以得到兩種晶體,金色塊狀的Bk(IO3)3與柱狀的Bk(IO3)4,其中后者是目前唯一得到單晶結構表征的Bk(Ⅳ)化合物[141]。在這兩種碘酸根配合物中,Bk(Ⅲ)和Bk(Ⅳ)分別處在扭曲的三帽三棱柱(九配位)與近乎標準的四方反棱柱(八配位)中心。顯微固態光譜儀顯示,Bk(IO3)3和Bk(IO3)4在熒光譜和吸收譜上存在顯著差異,進一步證實了其中锫具有不同的氧化態。
(3) 锎
目前普遍認為锎能以+2、+3、+4三種氧化態存在于固相中[142]。1968年,Cohen等[143]發現Cf(Ⅱ)可能存在于固相的證據。在加熱條件下,用氫氣處理含有Cf3+的EuCl3固體,產物溶于水后可與硫酸根離子形成可能為Eu(Cf)SO4的共沉淀。Mailen等[144]隨后也在熔鹽/液態金屬體系中發現了Cf(Ⅱ)存在的證據。Mikheev等[145]則發現,金屬鎂能在乙醇中還原Cf3+和Sm3+的混合氯化物,而生成的Cf2+能與SmCl2共結晶。后續的研究表明,SrB4O7基質也可用于穩定Cf(Ⅱ)[146]。1972年,Peterson等[147]在加熱條件下,用H2還原CfBr3,制備了首例純的Cf(Ⅱ)化合物CfBr2。在加熱條件下,CfBr3能自發分解產生CfBr2,并生成溴單質[148]。CfI2也可通過氫氣還原CfI3、或熱還原的方式制備[149]。盡管CfCl3的還原要困難得多,但仍有報道:在加熱條件下,用氫氣還原CfCl3可得到CfCl2,產物得到了固態光譜表征[142]。此外,Gibson等[150]在氫氣中加熱金屬锎,得到CfH2+x,并且發現在常規的合成條件下并不會形成CfH3。研究認為,锎在其氫化物中的真實價態可能介于+2到+3之間[151]。
Haire等[152]用氟氣處理CfCl3·xH2O、CfF3或Cf2O3得到了CfF4。三元氟化物MCfF5、M2CfF6、M3CfF7和M7Cf6F31(M為堿金屬)被預測能夠穩定存在,并且相比CfF4有著更高的熱穩定性[142]。Baybarz等[153]利用氧氣或鉑催化裂解出的原子氧對Cf(Ⅲ)進行氧化,發現在高溫下可以生成CfO2。锎的混價氧化物Cf7O12也曾被報道,它在750 ℃以上會逐漸失氧而轉化成Cf2O3[142]。
(4) 锿
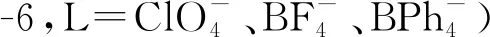
(5) 鐨
1972年,Mikheev等[158]發現,用鎂還原Fm3+和Sm3+的混合氯化物,得到的Fm2+可與SmCl2共結晶。隨后,他們在乙醇溶液中用Yb2+還原Fm3+,使生成的Fm2+與SrCl2共結晶,并通過實驗數據估算出E(Fm3+/Fm2+)=E(Yb3+/Yb2+)±0.02 V[159]。基于此,他們設計了一種基于Yb(Ⅱ)-乙醇-水溶液體系的Fm2+與NaCl共結晶的快速分離方法,可以在10 min內將鐨從锎、锿與鑭系元素等構成的混合物中分離,單次結晶步驟分離因子可達103至104量級[160]。TmI2可以在四氫呋喃(THF)中還原Fm(Ⅲ),產物與[Sr(18-c-6)]I2反應可得到[Fm(18-c-6)]I2[90]。此外,在LiCl-NdCl2-NdCl3熔鹽體系中,Fm(Ⅲ)和Es(Ⅲ)也可被還原至+2氧化態[160]。
(6) 鍆、锘、鐒
二價鍆、锘在固相存在的證據來自于共沉淀實驗。Hulet等[161]首先在BaSO4共沉淀實驗中確認了Md2+的存在。1979年,他們又用示蹤量256Md和SmF2/SmF3、SmCl2、RbCl或Rb2PtCl6等進行共沉淀反應,發現鍆離子具有和Fm2+、Eu2+、Sr2+相似的性質,而不同于Cs+。據此,他們認為其中鍆為Md2+,并否認了該條件下Md(Ⅰ)的存在[162](見4.4節)。锘的穩定二價形態是在LaF3或BaF2共沉淀反應中確定的[163]。對NoH2、LrH2等的理論計算表明,锘、鐒的化合物可能分別與鐳、鉈的化合物具有相似性[164]。
4 水溶液
4.1 釷和鈾
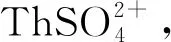
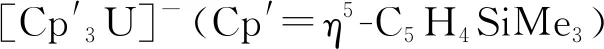
4.2 钚
Tananaev等[173]設計了一系列合成與反應性實驗,以確認Pu(Ⅷ)的真實性。比如,他們發現在2 mol/L NaOH溶液中,電解Pu(Ⅵ)也可以得到含有Pu(Ⅷ)的溶液。钚的(超)高價混合溶液可以將Np(Ⅵ)氧化至Np(Ⅶ),而這一氧化過程中既有Pu(Ⅶ)也有Pu(Ⅷ)的參與。不過,他們也指出,不能完全排除化學氧化或電化學氧化形成的“Pu(Ⅷ)”是Pu(Ⅶ)過氧化物的可能性[173]。Tsushima[174]利用DFT計算估計Pu(Ⅷ)/Pu(Ⅶ)的還原電勢,發現其值在+1.06 V到+4.36 V范圍內變化。其數值取決于所涉及物種的Pu-O鍵數目,以及Pu的配位數與配位模式,這突顯了高價钚溶液體系的復雜性。2014年,Kiselev等[175]發現,在溶液相中,離子型Pu(Ⅷ)物種可能與中性PuO4分子存在平衡。利用CCl4或CHCl3可以萃取水相中的PuO4,但此時七價物種仍然留在水相中。CCl4和CHCl3萃取液的吸收光譜表現出類似OsO4、RuO4等高價金屬氧化物的特征,其中332 nm處的最大吸收峰被指認為PuO4的π(O)→Pu荷移躍遷峰。利用XeF2的飽和CCl4或CHCl3溶液與PuO3·xH2O固體接觸,也能得到具有相似光譜學特征的Pu(Ⅷ)溶液。
4.3 其他前錒系元素
(1) 錒
1983年,Yamana等[176]研究了水溶液中Ac(Ⅲ)的放射極譜還原化學。通過向體系中引入18-c-6,半波電位有非常大的移動,這被歸因于形成了Ac(Ⅱ)-冠醚配離子。通過對離子半徑的估算,其中Ac2+的基態價電子構型被推測為6d1。考慮到Ac3+/2+的還原電勢估算值為-4.9 V(vs. NHE)[14],Ac(Ⅱ)能否在水溶液中存在尚需要更多表征結果予以確認。
(2) 鏷
Guillaumont等[177]通過理論計算發現,6d電子能穩定Pa(Ⅱ)但對Pa(Ⅲ)有去穩定化作用。因此,Pa3+、Pa2+的基態電子構型分別為5f2與5f26d1。對于Pa3+,其5f2構型與5f16d1構型之間能量差較小。因此,Pa3+可以較容易地從5f2基態轉化為5f16d1的激發態,隨后失去d電子而被氧化成Pa(Ⅳ)。據推算,Pa4+/3+的還原電勢為-2.1 V(vs. NHE),而Pa3+/2+的還原電勢則為-3.7 V至-4.4 V(vs. NHE)。
(3) 镅
在前錒系元素中,由于Am2+的半充滿5f7電子構型,镅的M3+/2+還原電勢負值最小(-3.7 V(vs. NHE))[178]。1976年,Pikaev等[179]對含有叔丁醇(氫氧根自由基的清除劑)的Am(ClO4)3水溶液進行脈沖輻解,并通過對產物的吸收光譜的分析得到了Am2+存在的證據。Sullivan[180]、Gordon[181]等同期報道了類似的結果,并展示了更詳細的輻解與動力學實驗過程。Shilov等[182]認為,輻解形成的Am2+在水溶液中重新被氧化的過程可能與Yb2+、Tm2+等相似,即經歷了水合離子的敏化、激基締合物的形成與分解等步驟。
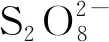
4.4 后錒系元素
(1) 鋦
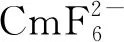
(2) 锫
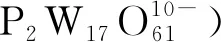
近年來,受益于毫克級249Bk生產技術的實現以及表征技術的進步,對溶液中Bk(Ⅳ)行為的研究得以深入[209]。2017年,Deblonde等[210]與合作者發現,在溫和條件下,由嗜鐵素衍生出的3,4,3-LI-(1,2-HOPO)配體(圖1)可以利用分子內的四個氮-羥基吡啶酮(HOPO)單元,實現對金屬中心的八齒螯合,從而穩定水溶液中的Bk4+。其中,Bk(Ⅳ)的價態得到了質譜、UV-vis吸收譜表征與DFT計算的支持。液相色譜-質譜(LC-MS)的實驗結果表明,這種仿生配體不僅可以實現對Bk(Ⅳ)的高度結合,還能有效地將Bk(Ⅳ)從含有Zr(Ⅳ)、Ce(Ⅳ)、Pu(Ⅳ)、Th(Ⅳ)等其他四價金屬離子的混合體系中分離出來[210]。
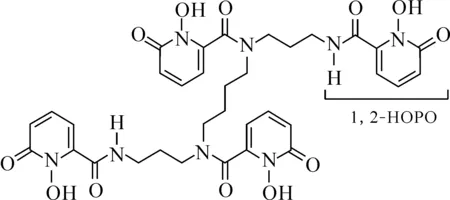
圖1 配體3,4,3-LI-(1,2-HOPO)結構[210]Fig.1 Structure of 3,4,3-LI(1,2-HOPO) ligand[210]
(3) 锎
Cf3+/2+的標準還原電勢據估算為-1.6 V(vs. NHE)[14]。1968年,Cohen等[143]利用硫酸鹽沉淀實驗給出了Cf2+在水溶液中存在的間接證據,他們發現Eu2+能還原Cf3+,但Cr2+不能。Gunningham等率先嘗試了水溶液中Cf3+的電化學還原,但由于窗口設置的問題,并沒有觀察到Cf2+的形成[211]。隨后,Friedman等[211]的放射極譜還原研究給出了Cf2+存在的間接證據。1981年,Musikas等[212]發現了水溶液中Cf(Ⅲ)在滴汞電極上的分步還原。不久后,Sullivan等[213]通過Cf(ClO4)3水溶液的脈沖輻解研究給出了Cf2+溶液的時間分辨吸收譜。Friedman等[211]還在乙腈中探究過Cf(ClO4)3的放射極譜還原,發現Cf(Ⅲ)和Sm(Ⅲ)有十分相似的還原性質。2019年,Marsh等[214]在THF中研究了锎-穴醚體系的還原化學,發現穴醚的引入提升了Cf(Ⅱ)的相對穩定性,還觀察到[Cf(crypt)]3+(crypt=穴醚[2.2.2])的電化學行為與Sm3+和Yb3+的對應配離子有相似之處。相比于還原,Cf3+的氧化難度更大[215]。Cf4+/3+的還原電勢估算值為+3.2 V(vs. NHE)[14],故時至今日仍然鮮有關于Cf(Ⅳ)溶液化學的研究[188]。Payne等[204]曾經報道在乙腈溶劑中三苯基氧胂可以穩定Cf(Ⅳ),但缺少足夠的表征結果來證實Cf(Ⅳ)的存在。
(4) 锿、鐨、鍆、锘、鐒
超锎元素的正四價離子尚無在溶液中存在的證據[188],其形成難度從Es4+/Es3+高達+4.5 V(vs. NHE)的還原電勢可見一斑[14]。因此,即便是HOPO類配體也很難穩定Es(Ⅳ)[216]。锿、鐨、鍆的M3+/2+的還原電勢估算值分別為-1.3、-1.2、-0.15 V(vs. NHE),而No3+/2+的還原電勢驟升至+1.45 V(vs. NHE),這是由于No2+具有全充滿的5f14殼層結構[14]。
Es2+、Fm2+的溶液化學研究均與電化學測量相關。比如,在硫酸根離子存在的條件下,水溶液中Es3+的極譜還原研究給出了Es(Ⅲ)→Es(Ⅱ)→Es(0)的分步還原機理[217-218]。鍆是第一個被發現能以正二價形態在水溶液中保持穩定的錒系元素。鋅粉、鋅汞齊以及Cr2+、Eu2+、Yb2+等均能將Md3+還原成Md2+,而V3+/2+電對能在水溶液中與Md3+/2+電對實現氧化還原平衡[161, 219]。流動電解色譜法測得的Md3+/2+還原電勢為(-0.16±0.05) V[220]。1976年Mikheev等[221]宣稱在乙醇中,用Eu2+或Yb2+可以將Md3+還原成5f14電子構型的Md+,并用CsCl或RbCl實現了Md+的晶格載帶。這一發現隨后遭到了Samhoun與David等的質疑,他們仔細重復了Md3+的電化學實驗,認為在Md2+→Md0的還原過程中并沒有Md+的生成[222-223]。對“Md(I)是否存在”這一問題學術界一直存有爭論[17, 90, 224-226]。
與Md(I)同樣具有5f14電子構型的No2+,則是锘在水溶液中最穩定的離子形態。這一特性使得最早宣稱合成102號元素的瑞典斯德哥爾摩大學團隊,在利用其發展的基于三價離子分離技術的錒系純化工藝時,無法獲得102號元素(锘)的純品[227]。Maly等[163]最早發現No2+和堿土金屬離子在色譜、共沉淀性質上的相似之處,而用Ce4+氧化之后得到的No3+與其他三價錒系金屬離子則有相似性質。No2+的結構化學與配位化學隨后得到了研究[228-229],而流動電解柱色譜技術的發展則為調控锘的氧化態提供了可能[230-231]。對于鐒而言,由于可供應核素較短的半衰期,僅有幾例嘗試還原Lr3+的研究被報道。但是,目前在水溶液中還沒有發現低價鐒離子(如Lr2+)存在的證據[232-234]。
5 總結與展望
近年來,錒系元素的非尋常氧化態化學的研究已經取得了重大突破。舉例來說,在1967年以前,人們對錒系元素+2氧化態的了解僅限于CaF2基質中摻雜的Am(Ⅱ),而六價以上的氧化態則尚未涉足[235-236]。到了1980年代后期,人們利用實驗結合理論的方法估算了幾乎所有錒系元素(除了鐒)的M4+/3+和M3+/2+的還原電勢值,并對七價镎、钚的合成與化學性質有了初步的了解[237]。在當下,無論在溶液相還是固相中,Pu(Ⅶ)和Np(Ⅶ)均已不再罕見[98, 106];而二價錒系金屬在不同相中的制備方法與理化性質也得到了較為深入的研究[90, 238-239]。
展望未來,錒系元素的非尋常氧化態化學仍有著廣闊的發展前景。在氣相,利用稀有氣體基質的穩定作用[240],發展錒系金屬-金屬鍵物種的合成化學一直是令人興奮的研究方向[34]。在固相,在稀土金屬相關研究中發現的MX、M2X3、M5X8、M6X7、M7X10、M7X12(M=Y、La、Ce、Pr、Gd、Tb、Er、Ho、Lu)等鹵化物簇有著重要的參考價值[90],為低價錒系金屬固體化學的進一步發展提供了新的可能。對于溶液化學,離子液體由于其寬闊、可調的電化學窗口而成為發展錒系元素氧化還原化學的潛力介質[241]。曾有關于U(Ⅳ)、Th(Ⅳ)化合物在離子液體中的電化學還原的報道,但認為其過程分別為U(Ⅳ)→U(Ⅲ)→U(0)、Th(Ⅳ)→Th(0),并未經歷U(Ⅱ)或Th(Ⅲ)、Th(Ⅱ)的形成[242-243]。對于后錒系元素而言,化學家們現在最主要的目標是,通過一系列對比研究,揭示結晶化合物中金屬中心的基態電子構型,由此反映后錒系元素周期性的轉變。在此基礎上,比較與探索錒系元素和同樣由正三價主導的鑭系金屬在成鍵性質和化學行為上的差異[151, 209, 244],從而實現核燃料后處理過程中鑭-錒分離等關鍵過程的優化。
綜上,錒系元素的非尋常氧化態研究無疑是當前錒系元素配位化學中最富生機、最具潛力、同時也最具挑戰的領域之一。合成化學在這一領域的發展中起到了關鍵性的作用,但仍需其他方向上的協作才能取得更大進展。尤其是對于超钚元素來說,核素生產能力的提升[245-246]、傳統測試方法的改良以及新表征技術的引入[209],是其配位化學研究在最近幾年接連取得突破的先決條件。比如,2000年后,核素249Bk(T1/2=320 d)實現了毫克級的批量制備,這一技術突破極大地加速了锫的化學研究進展。又如,Galbis等[247]在2010年首次將擴展的X射線吸收精細結構(EXAFS)譜學方法與蒙特卡羅模擬相結合,解決了Cf3+水合離子結構的分辨問題。這推動了EXAFS在超钚元素溶液化學研究中的大量應用[244]。而結合磁圓二色譜(MCD)、電子順磁共振(EPR)、超導量子干涉儀(SQUID)等表征手段,也將極大促進對錒系元素非尋常氧化態的電子結構、磁學特征的進一步了解。未來,錒系元素非尋常氧化態的研究將不僅限于基礎研究,也將在諸多應用領域,如核燃料后處理、單分子磁體等方面展現巨大潛力[248]。通過更廣泛、更深入的交流與合作,我們期待并堅信中國學者和中國團隊會在錒系元素的非尋常氧化態化學和相關學科交叉領域做出更多的貢獻。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
科技知識動漫(2017年7期)2017-08-09 19:52:45
汽車工程學報(2017年2期)2017-07-05 08:13:02
科技知識動漫(2017年5期)2017-05-11 21:34:16
科技知識動漫(2017年4期)2017-04-15 22:24:55
科技知識動漫(2017年2期)2017-02-06 20:59:46
