談GMP文件體系及其管理
2021-01-11 16:14:21冼海燕
中國藥學藥品知識倉庫 2021年17期
冼海燕
摘要:良好的文件是質量管理體系的重要組成部分,是符合藥品生產質量管理規范(GMP)要求的關鍵。GMP文件體系中的各項文件應包括充分的指導細節,以便于執行者對標準和要求有著相同的理解;同時還應提供對各種過程的充分記錄,以便證明既定的標準和要求是持續被遵守的。
關鍵詞:GMP文件體系;文件管理
【中圖分類號】 R-1? ? 【文獻標識碼】 B? ? ? 【文章編號】2107-2306(2021)17--01
GMP,是“藥品生產質量管理規范”的Good Manufacturing Practice英文名稱簡稱。GMP(2010年修訂版)第一百五十條指出:“文件是質量保證系統的基本要素。必須有內容正確的書面質量標準、生產處方和工藝規程、操作規程以及記錄等文件。”也就是說,按規定建立GMP文件體系,用通俗的行話講,“寫你做的,做你寫的,記你做的”。主要目標是建立、控制、監測和記錄直接或間接影響藥品質量各方面的所有活動,以確保藥品生產的各項活動持續遵守既定的標準和要求。那么,GMP文件體系包括哪些內容及其要求又有哪些?
1.GMP文件體系制定時的一般原則包括:
1.1所有可能影響藥品特性、質量的生產、包裝、存儲、銷售和實驗室的操作,以及執行GMP的輔助性文件(如清潔記錄、預防性維護報告等)都必須有書面文件,且由質量管理部門審核和批準。
1.2GMP文件必須符合GMP、中國藥品管理法等法規、規范的要求。
1.3建立GMP文件體系前,必須明確與文件制定、批準和使用相關的角色和及其職責。文件審核過程必須確保文件準確、完整、符合法規要求,所下的結論是有科學依據的。
1.4起草、審核和批準GMP文件的人員必須對相應的設備和工藝有適當的技術理解。
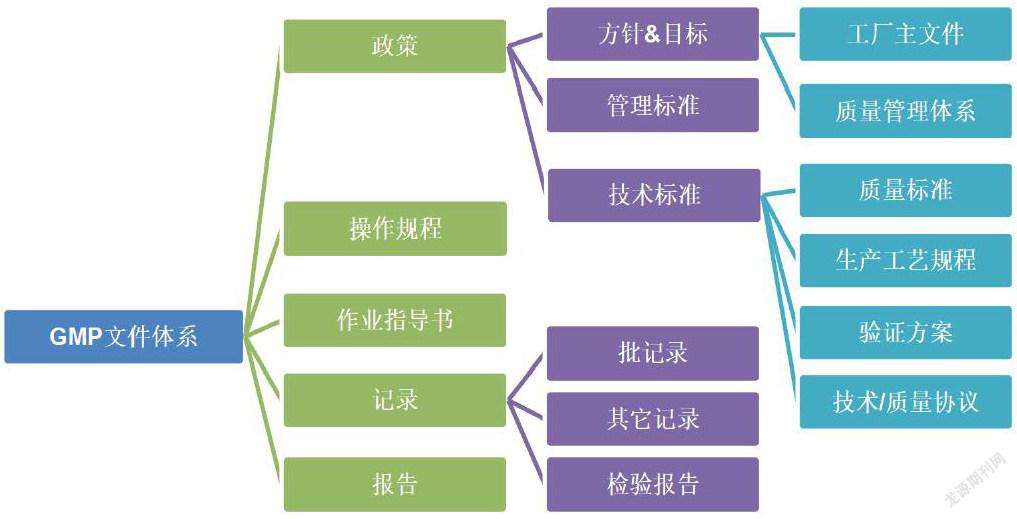
2.GMP文件體系的構成
2.1建議GMP文件體系有以下文件:
2.1.1工廠主文件:描述工廠與執行GMP相關的所有活動。
2.1.2質量管理體系,是以文件化的形式建立了實現有效質量管理所需的組織結構、職責和規程的正式體系。質量管理體系至少應建立確保產品質量和患者安全所需的文件,包括但不限于政策、程序或記錄,例如:
·人員和培訓
·文件管理
·生產和過程控制
·標簽和包裝控制
·來料驗收
·糾正和預防行動
·變更管理
·質量審計
·過程質量監控
·儲存和發運
·投訴處理
·年度報告
2.1.3指引類文件:包括技術標準、管理規程和操作規程,例如:
2.1.3.1質量標準:詳細描述生產過程中使用或制得的產品或物料必須符合的要求。
2.1.3.2生產處方和工藝規程:提供所有要使用的起始原料、設備和計算機系統的詳細信息,并說明所有工藝、包裝、取樣和檢驗操作說明。同時,應規定要采用的過程控制和過程檢驗技術以及可接受標準。
2.1.3.3操作規程&作業指導書,例如確認和驗證、校準、設備的維護及清潔和消毒、培訓、更衣、環境監測、蟲害控制、偏差處理、藥品召回、退貨等。
2.1.3.4方案:給出執行和記錄某些特定操作的說明,如驗證方案。
2.1.3.5技術/質量協議:由委托方和受托方就GMP外包活動達成的協議。
2.1.4記錄和報告
2.1.4.1記錄:提供各項活動符合操作說明的證據,例如活動、事件、調查。記錄還包括用于生成其它記錄的原始數據。
2.1.4.2批記錄:是記錄每批產品的歷史記錄。
2.1.4.3檢驗報告書:提供產品或物料樣品的檢驗結果及結論。
2.1.5報告:記錄特定活動、項目或調查的進行情況,以及結果、結論和建議。包含不僅限于以下內容:
·多產品共用的可行性評估報告
·確認或驗證報告
·持續穩定性考察報告
·現場質量審計報告
·定期的質量回顧分析報告
·召回報告
·GMP自檢報告
·年度報告
2.2 GMP文件體系
3.GMP文件的控制
3.1應有文件定義文件類型,并且有文件管理規程。
3.2隨著科學技術的發展和應用,許多文件如操作規程或作業指導書和記錄等,可能以電子文件和紙質文件的形式共存。此類情況,應說明原文件、正式復制件、數據處理和記錄的關系及控制措施。
3.3應制定控制措施對電子文件實施適當的控制,以確保記錄在整個保存期內的完整性。
3.4 GMP變更控制規程應包含GMP文件的修訂,該變更必須由經授權的人員進行。
3.5 GMP文件變更控制包括:
3.5.1控制所有文件的發布、修訂、替換和撤銷,以確保提供和使用正確的文件。
3.5.2修訂歷史的使用和維護。
3.5.3在使用新修訂的文件之前,必須對該文件進行培訓并記錄。
3.6如使用電子數據處理系統、照相技術或其他可靠方式記錄數據資料,應當有所用系統的操作規程;記錄的準確性應當經過核對。
3.7使用電子數據處理系統的,只有經授權的人員方可輸入或更改數據,更改和刪除情況應當有記錄;應當使用密碼或其他方式來控制系統的登錄;關鍵數據輸入后,應當由他人獨立進行復核。
4. 良好文件規范
4.1所有記錄的手寫內容必須清晰,并用不褪色的筆書寫。
4.2 GMP文件中的簽名必須清晰可辨,以便可追溯到執行該步驟的人員。在適當的情況下,電子簽名可以代替手寫簽名。
4.3記錄應當保持清潔,不得撕毀和任意涂改。
4.4應遵照數據可靠性原則。
4.5在執行每個操作和/或發生需要記錄的事件時都應填寫記錄。必須根據相應的 GMP 文件的職責對記錄的內容進行檢查、確認、審核和/或批準。
4.6必須管理和控制記錄原件,以確保將不當使用和偽造記錄的風險降低到可接受的水平。
5.數據可靠性原則
在數據的整個生命周期中,記錄必須是可追溯的、清晰的、及時的(在發生時記錄)、原始的和準確的(ALCOA)。
5.1可追溯的(Attributable):應能確定執行記錄任務的人。需記錄誰執行了任務、誰對記錄做了更正、刪除、更改等。
5.2清晰的(Legible):所有的記錄都必須是清晰易讀的。
5.3及時的(Contemporaneous):應在行動、活動或決策發生時進行記錄。
5.4原始的(Original):原始記錄可以描述為第一次獲取的信息,無論是紙質記錄還是電子記錄。
5.5準確的(Accurate):通過諸多因素的實施能確保結果和記錄的準確性。
6.GMP文件的保存
6.1必須保存所有GMP文件。
6.2文件必須以安全、可靠的方式保存,并可快速檢索。
6.3記錄必須完整。
6.4記錄必須僅供授權的用戶查閱或訪問。
6.5在要求的保存期內必須保證GMP文件或記錄的數據的完好性和完整性。
6.6批記錄的保存期限至少按照監管規范的規定執行;而其它類型的文件的保存期限,取決于文件所支持的業務活動,包括在上市許可有效期內,保留作為上市許可信息支持的原始數據(例如與驗證或穩定性相關的數據)。
6.7文件的保存期限應在內部操作規程中規定。
【討論】
良好的文件是質量管理體系的重要組成部分,是符合藥品生產質量管理規范(GMP)要求的關鍵。GMP文件體系中的各項文件應包括充分的指導細節,以便于執行者對標準和要求有著相同的理解;同時還應提供對各種過程的充分記錄,以便證明既定的標準和要求是持續被遵守的。
參考文獻:
[1] 藥品生產質量管理規范(2010年修訂)(衛生部令第79號) 2011.1.17發布
[2] MHRA.GMP Data Integrity Definitions and Guidance for Industry.Jan,2015
[3] FDA.Data Integrity and Compliance with CGMP Guidance for Industry.Apr,2016
[4] Model of Organization, Management and Control, referred to in the Italian Legislative Decree 231/01 and subsequent amendments.
猜你喜歡
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
中國衛生(2016年5期)2016-11-12 13:25:28
汽車觀察(2016年3期)2016-02-28 13:16:26
中國衛生(2015年5期)2015-11-08 12:09:48
中國衛生(2014年7期)2014-11-10 02:33:02
新高考·高一物理(2014年1期)2014-09-18 01:26:07
