海洋來源活性肽及其抗炎活性研究進展*
2021-01-14 08:44:44龍潔怡劉永宏
廣西科學 2020年5期
楊 斌,龍潔怡,2,劉永宏,2,3**
(1.中國科學院南海海洋研究所,中國科學院熱帶海洋生物資源與生態重點實驗室,廣東省海洋藥物重點實驗室,廣東廣州 510301;2.中國科學院大學,北京 100049;3.廣西中醫藥大學,廣西南寧 530200)
0 引言
炎癥是免疫系統對外部挑戰或組織損傷的一種普遍、復雜和有益的生理反應,可以恢復組織結構和功能。炎癥反應的發生有助于傷口治愈,但當炎癥過激或不可控時,異常而長時間的炎癥反應將引發多種疾病。由免疫細胞活性氧物質引起的基因突變,以及炎癥反應中涉及的炎癥介質,可能會引發許多疾病,例如癌癥、多發性硬化、動脈粥樣硬化、關節炎、心臟疾病、胰島素抵抗等[1]。因此,研究抑制炎癥介質的過度產生是治療這些疾病的有力策略。目前臨床常用的抗炎藥主要有甾體類抗炎藥、非甾體抗炎藥和生物制劑,但這些藥物都有一定的副作用,所以在臨床使用上受到限制。
海洋生物為了更好地適應極端的生存環境,形成獨特的遺傳系統和生物合成途徑,并產生大量結構新穎、生物活性豐富的次級代謝物[2],為藥物開發開辟了新的前景,越來越多的候選藥物進入臨床試驗或已批準上市[3]。其中,多肽因具有獨特結構、活性高、分子量小、靶向性強、毒性低、易于跨膜吸收等特點被廣泛研究,并且許多化合物已被證實具有抗真菌、抗病毒和抗增殖等作用[4]。但迄今為止從海洋棲息地中發現抗炎肽的例子還很少,海洋來源的具有特殊細胞靶標的新型生物活性肽的發現,可能有助于尋找有前途的先導藥物或候選藥物[5,6]。本文綜述了自20世紀90年代以來,具有治療炎癥相關疾病潛力的海洋來源活性肽的研究進展,為開發新的抗炎藥物提供參考。
1 炎癥信號途徑及抗炎活性研究模型
海洋來源環肽類抗炎化合物多數作用于NF-κB信號途徑,該途徑是經典的炎癥信號途徑[7]。位于細胞質中的NF-κB由兩個亞基(p50和p65)組成,它們是抑制蛋白IκB-α的非活性異二聚體。在刺激條件下,IκB-α的磷酸化和蛋白水解使NF-κB易位進入核內,然后通過與DNA結構中的κB位點結合來調節靶基因的轉錄[8]。NF-κB的反式激活會增加下游炎癥介質的表達,例如IL-1β、IL-6和TNF-α等促炎細胞因子[9],重要的促炎酶(例如誘導型一氧化氮合酶iNOS和環氧化酶COX-2)及其派生產物NO和PGE2[10]。炎癥介質的過度產生引起細胞損傷,例如發紅、疼痛、發燒和腫脹[11]。因此,抑制炎癥介質的過度產生是治療炎癥性疾病的重要目標,可以作為評估藥物發揮抗炎作用的指標。除NF-κB活化外,還有一種涉及細胞外信號調節激酶(ERK)、p38和cJun NH2末端激酶(JNK)的MAPK信號途徑,也可以激活炎癥,并調節各種炎癥相關基因的轉錄,調節炎癥因子的基因表達[12]。
研究人員常用的體外抗炎活性研究模型有兩種:(1)建立脂多糖(Lipopolysaccharides,LPS)、佛波酯(Phorbol 12-myristate 13-acetate,PMA)或其他藥物刺激的體外免疫單核細胞和巨噬細胞(BMDM細胞、RAW264.7細胞等)等體外模型[13]。通過這些模型評價藥物對一氧化氮釋放的影響,以及ELISA、Western blotting分析相關炎癥因子(IL-1、IL-6、TNF-α、PGE2等)和蛋白( iNOS和COX-2等)表達水平,是判斷天然產物是否具有潛在抗炎活性的有效手段[14]。(2)通過LPS刺激中性粒細胞,檢測中性粒細胞彈性蛋白酶(Enutorphil Elastase,NE)的表達,NE抑制劑抑制NE活性,調節炎性細胞因子和趨化因子的釋放,抑制炎細胞激活、跨膜遷移及組織毒性物質釋放[15],發揮多層次的抗炎效應。在體內實驗也主要有兩種研究模型:(1)在大鼠足跖內注射卡拉膠能使大鼠出現局部毛細血管擴張、血管通透性增高、組織液滲出、足水腫等一系列類似于人體急性炎癥的反應[16]。(2)二甲苯或PMA誘導小鼠耳腫脹,然后通過檢測炎癥部位改善情況評估抗炎效果[17]。
2 海洋來源抗炎活性肽分子
海洋環境復雜多變,生物種類豐富,蘊含著大量結構新穎的活性化合物,肽類為其中一類重要的活性天然產物,是未來藥物開發的方向之一。目前為止,已發現的具有抗炎作用的海洋來源活性肽主要有4類。
2.1 環肽類及其抗炎機制
Gulavita等[18]從澳大利亞西南部采集到的海綿Theonellasp.中分離得到環八肽perthamide B (1),化合物1顯示輕微的抑制細胞因子IL-1β表達,半抑制濃度(50% Inhibitory Concentraton,IC50)為27.6 μmol/L。Festa等[19]從所羅門群島的海綿Theonellasp.中發現perthamides C—K (2—10),其中perthamides C (2)和D (3)具有顯著的抗炎活性;在卡拉膠誘導的小鼠足水腫模型中,化合物2,3,8,7和10均能顯著降低卡拉膠誘導的足水腫,且無論在早期階段(0—6 h)還是在晚期階段(24—96 h),其作用程度呈劑量相關,當化合物2腹腔注射(ip)用藥劑量為300 μg/kg時能降低60%小鼠足水腫。佛波酯(PMA)能誘導死后人腎(PHK)細胞引起的末端分化和炎癥反應,是研究銀屑病常用的體外模型之一[20]。perthamide C (2)和perthamide E (4)能抑制組織型纖溶酶原激活劑(Tissue Plasminogen Activator,TPA)誘導PHK細胞炎癥因子上調,化合物4能抑制IL-8表達,而化合物2能同時抑制TNF-α和IL-8的表達起抑制炎癥作用,提供新的抗銀屑病研究方向[21]。當perthamide G (6)中的脯氨酸替代化合物2的γ-甲基脯氨酸,抗炎作用消失;具有ADAA殘基的perthamide J (9)具有強抗炎作用。研究表明同時擁有γ-甲基脯氨酸、AHMHA和ADAA結構片段,是perthamides化合物分子產生抗炎作用的藥效條件[4]。
以上化合物的化學結構見圖1。
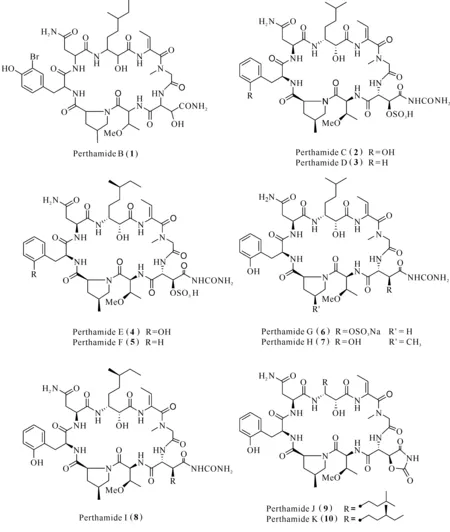
圖1 化合物perthamides (1—10)的結構
Cyclomarins A (11)、B (12)和C (13)是從海洋放線菌Streptomycessp.提取得到的環七肽(圖2);cyclomarin A由3種常見氨基酸和4種特殊的氨基酸組成,在體內與體外實驗都表現出顯著的抗炎作用;在PMA誘導小鼠耳腫脹模型中,cyclomarin A在50 μg/耳的劑量下耳腫抑制率達92%;腹腔注射給藥劑量30 mg/kg時,耳腫抑制率達45%[22],表明該化合物是一種潛在的抗炎候選藥物。
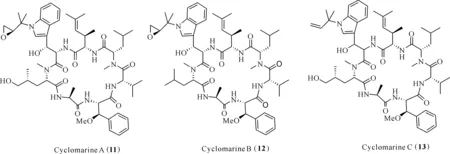
圖2 化合物cyclomarins (11—13)的結構
Solomonamides A (14)和B (15)是來自海綿Theonellaswinhoei的環肽新骨架(圖3),化合物14能夠降低卡拉膠誘導的足水腫,以100μg/kg(ip)的劑量能使抑制率達60%,呈劑量相關[23]。
來自海綿Stylissamassa的新天然產物 stylissatin A(16)[24]及其全合D-allo-Ile4叔丁基衍生物(17)[25](圖3),在LPS誘導的RAW264.7細胞炎癥模型中,能顯著抑制一氧化氮的生成,IC50分別為87和12 μmol/L。
IL-10作為免疫抑制因子,通過抑制多種效應分子來抑制機體的抗腫瘤免疫,通過誘導產生IL-2和IFN-C來抑制IL-12及TNF-α的表達,同時作為促炎細胞因子參與TNF-α的負反饋調節。Liu等[26]從海綿共附生真菌Aspergillusviolaceofuscus中發現環四肽violaceomide A (18)(圖3),在LPS誘導的THP-1源巨噬細胞中,當用藥濃度在10 μmol/L時,化合物18顯著抑制促炎細胞因子IL-10的表達,抑制率為84.3%。
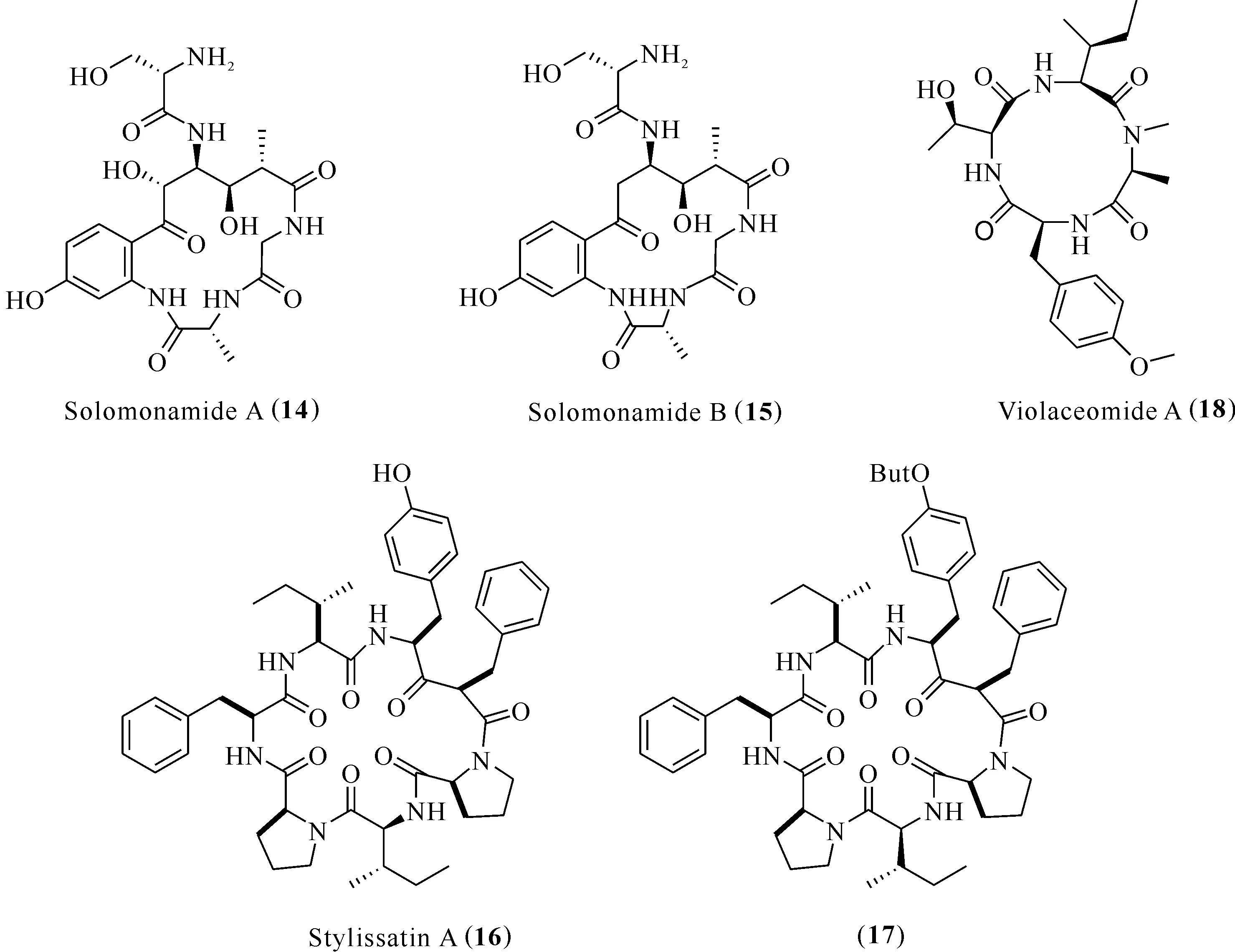
圖3 化合物Solomonamides (14,15)、stylissatin A (16)和(17),violaceomide A (18)的結構
Nalli等[27]從海洋鏈霉屬Streptomycessp.中分離獲得10個吡嗪-1,4-二酮取代環二肽類化合物(19—28)(圖4),其中化合物21,22,23,25和26對脂多糖誘導的巨噬細胞中的兩種促炎細胞因子TNF-α和IL-6均具有良好的抑制作用,在10 μmol/L用藥劑量下,抑制率為23.7%—62.1%;體外和體內實驗都證實化合物25是TNF-α的特異性抑制劑,可有效抑制LPS誘導的外周血單個核細胞(PBMCs)中TNF-α的產生。
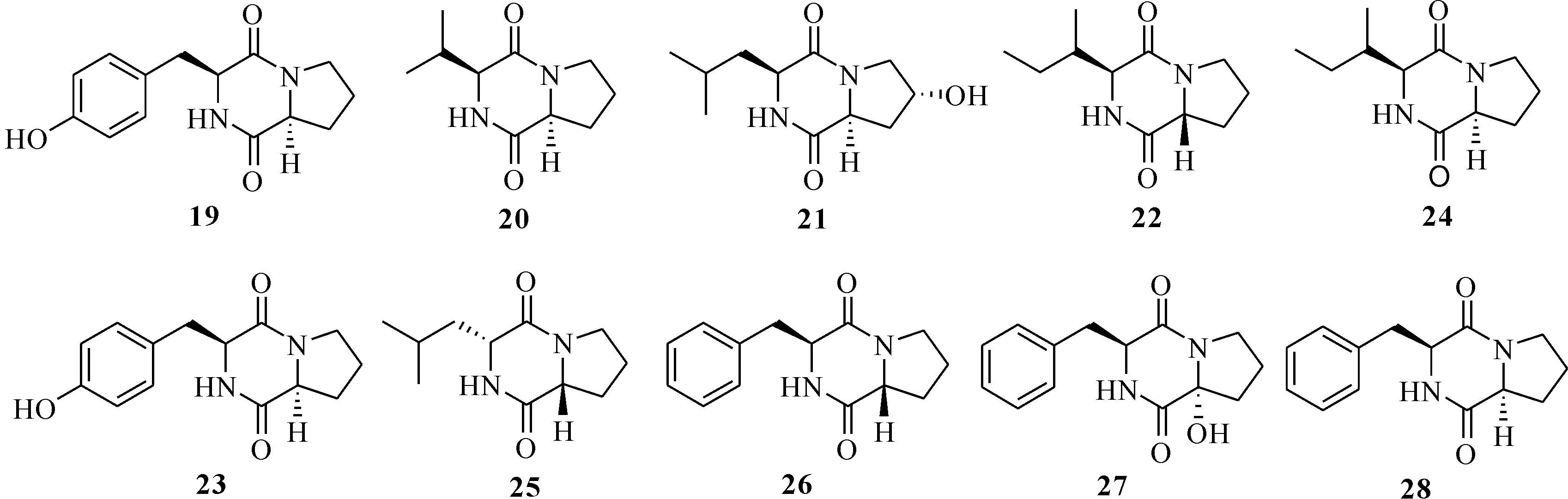
圖4 吡嗪-1,4-二酮取代環二肽類化合物(19—28)的結構
2.2 縮肽類及其抗炎機制
縮肽化合物是一類包含酯鍵的多肽,是羥基酸和氨基酸由酯鍵和酰胺鍵連接構成的一類寡聚物,具有抗菌、抗病毒、抗血栓、抗蟲以及抗腫瘤等多種生物活性[28]。
TPA誘導小鼠耳腫是一種模擬非特異炎癥的動物模型,TPA誘導小鼠耳組織角質形成細胞產生TNF-α、IL-1β和IL-6等促炎細胞因子[29]。從水母表面共附生放線菌Streptomycessp.中提取得到的salinamides A—E (29—33)(圖5),其中salinamides A (29)和B (30)具有較好的抗炎作用,在TPA誘導的小鼠耳腫模型下,以50 μg/耳的給藥劑量,抑制率分別達84%和83%[30]。
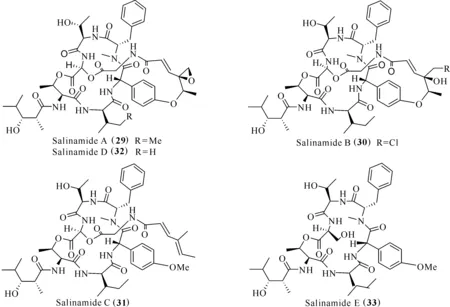
圖5 化合物Salinamides (29—33)的結構
Halipeptins A—C (34—36)[31]是從海綿Haliclonasp.提取獲得的17元環縮酯肽(圖6),化合物34具有較好的抗炎特性,在體內的抗炎作用比商業抗炎藥物如萘普生和吲哚美辛更強。在小鼠皮下氣囊(Subcutaneous Air Pouch,SAP)慢性炎癥模型中,通過足下注射卡拉膠誘發炎癥反應,經藥物處理后發現,在300 μg/kg給藥劑量下,能減輕60%足水腫[32]。

圖6 化合物halipeptins (34—36)的結構
Thalassospiramides A (37)、B (38)、D (39)和G (40)[33]分離自海洋細菌Thalassospirasp.(圖7),化合物38對Th2細胞免疫應答中上調的IL-5產生抑制作用,IC50為5 μmol/L[34]。進一步研究發現,thalassospiramides A (37)和D (39)能抑制LPS誘導的RAW264.7巨噬細胞中一氧化氮的釋放,IC50分別為16.4和4.8 μmol/L[35]。
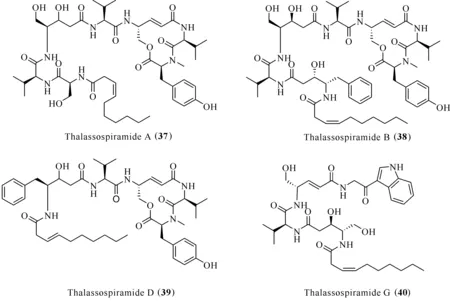
圖7 化合物thalassospiramids (37—40)的結構
從深海鏈霉菌SomaliensisSCSIOZH66中提取分離的抗霉素型縮肽somalimycin (41)、USF-19A(42)和urauchimycin D (43)(圖8),能夠抑制卵白蛋白誘導哮喘小鼠脾細胞中IL-5的釋放。化合物42的IC50達0.57 μmol/L,化合物41和43的IC50均大于10 μmol/L,但細胞毒半數致死量LD50僅為34.6—192.9 μmol/L,具有良好的抗炎潛力[36]。

圖8 化合物Somalimycin (41)、USF-19A (42)和urauchimycin D (43)的結構
2.3 脂肽類及其抗炎機制
脂肽類是一類結構復雜、生物活性顯著的化合物,具有抗細菌、抗真菌、抗腫瘤、抗炎[35]等生物活性。脂肽類化合物的應用前景較為廣泛,具有較大的開發價值。
Microcolin A (44)、microcolin B (45)和microcolin D (46)是從藍藻Lyngbyamajuscula中分離得到的(圖9)[37]。淋巴細胞功能相關抗原LFA-1是白細胞整合素家族的一員,參與T細胞與抗原呈遞細胞的結合。胞內黏附分子ICAM-1是胞內黏附分子的一種類型,在細胞與細胞互相作用時,抑制白細胞浸潤。LFA-1/ICAM-1相互作用在炎癥疾病的發病機制中發揮重要作用[38]。化合物45和46抑制LFA-I/ICAM-I介導的細胞黏附,IC50分別為0.15和0.9 μmol/L,有望被開發為LFA-I/ICAM-I抑制劑[39,40]。化合物44能夠阻止IL-2的產生與IL-2受體的表達,并且對刀豆凝集素A (Concanavalin A)活化小鼠脾細胞淋巴細胞的增殖有抑制作用[41]。
Malyngamides類化合物分子中的脂肪酸鏈與具有環己烯酮或環氧環己酮結構的胺均通過酰胺鍵連接,是一類骨架新穎、結構復雜的脂肪酰胺化合物,具有良好的生物活性[42]。Malyngamide S (47)能抑制分別由趨化肽(fMLP)和PMA誘導的人中性粒細胞中超氧陰離子的生成,抑制率分別為73%和83%[43]。Malyngamide C (48)、malyngamide J (49)和malyngamide L (50)分離自藍藻Lyngbyamajuscula,能抑制LFA-I/ICAM-I介導的細胞黏附,伴隨著較小的細胞毒性[39]。Malyngamide 2 (51)分離自未知藍藻,能夠抑制LPS誘導的RAW264.7巨噬細胞一氧化氮的釋放,IC50為8.0 μmol/L。
以上化合物的化學結構見圖10。
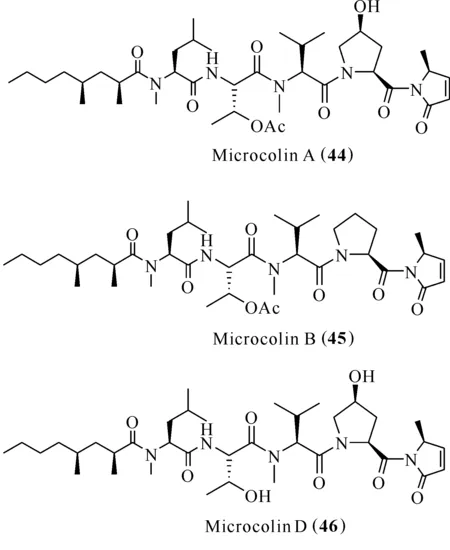
圖9 化合物Microcolins (44—46)的結構
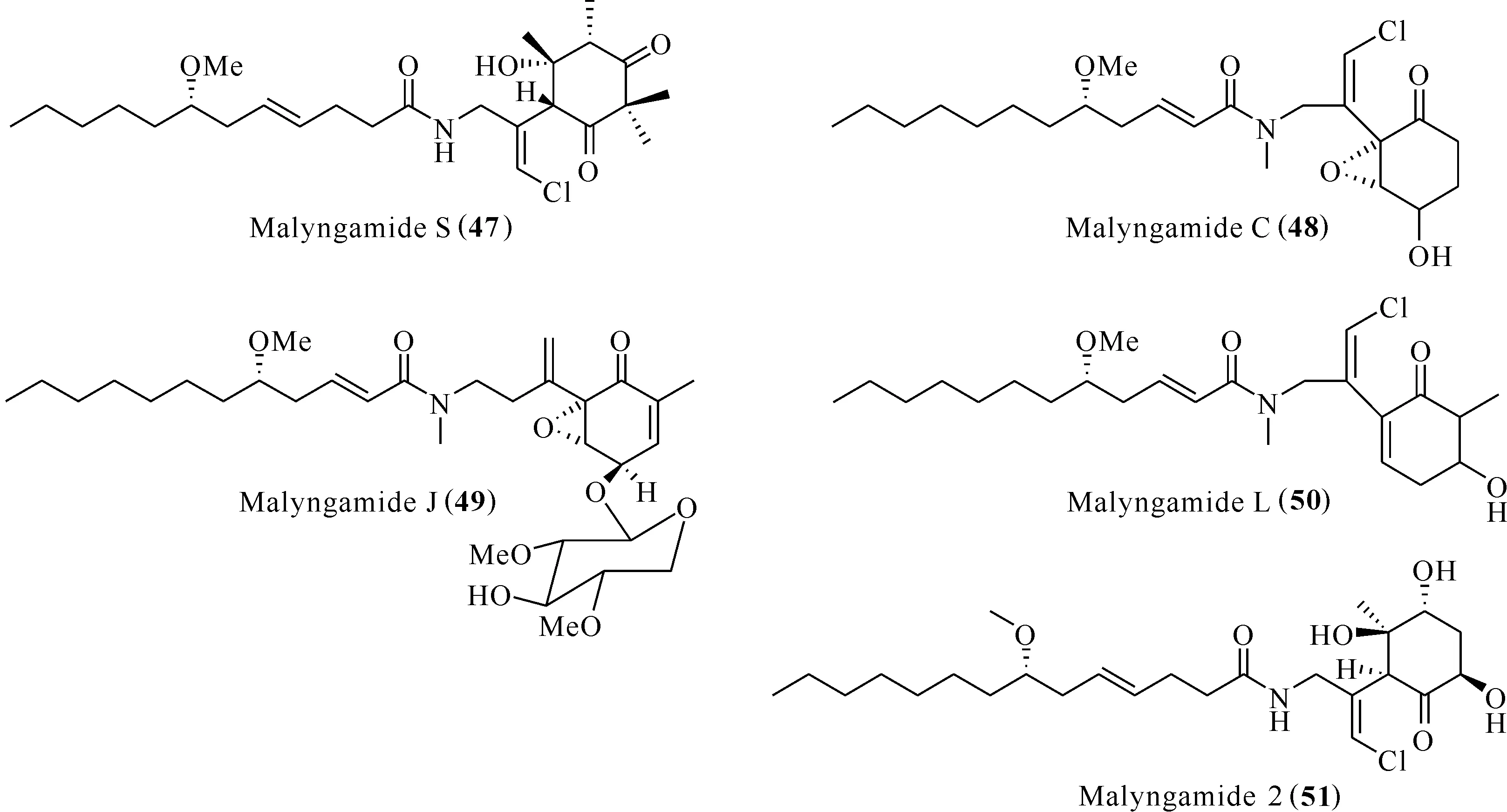
圖10 化合物Malyngamides (47—51)的結構
2.4 蛋白質類及其抗炎機制
藻藍蛋白(Phycocyanin,PC)是一種光合色素,廣泛存在于藍藻和紅藻細胞內,其分子量在40 kU左右,由α和β兩個亞基組成,肽鏈上共價結合1個開鏈的四吡咯環輔基。藻藍蛋白在小鼠模型中被證明可以抑制各種類型的水腫和活性自由基的釋放,類似于其他非甾體抗炎藥,如吲哚美辛和布洛芬[43]。在花生四烯酸(AA)誘導的小鼠耳炎模型中,藻藍蛋白可以抑制前列腺素E2的產生、抑制磷脂酶A2的活性和組胺的釋放[44-46]。進一步的研究發現藻藍蛋白能抑制COX-2蛋白的表達和促炎細胞因子TNF-α的釋放[47]。
凝集素(Lectin)是一種糖蛋白,分離自各種植物、無脊椎動物和高等動物。最近的研究發現,海洋生物中也存在凝集素,并且具有一定的抗炎作用。在酵母聚糖誘導的顳下頜關節骨關節炎動物模型中,預先用分離自綠藻Caulerpacupressoides的海藻凝集素保護,隨后用酵母聚糖誘導炎癥,結果發現其可降低小鼠關節痛并抑制促炎細胞因子IL-1β和TNF-α的生成,并抑制白細胞浸潤以及降低髓過氧化物酶(MPO)的活性。在卡拉膠誘導小鼠足水腫模型中,該凝集素能顯著降低腫脹率和中性粒細胞的滲透,也可以抑制促炎細胞因子IL-1β、TNF-α、IL-6和COX-2表達。除此之外,海藻凝集素對葡聚糖和組胺誘導的足水腫也有抑制作用,但對5-羥色胺和緩激肽誘導的足水腫沒有效果,表明該凝集素的抗炎作用可能與抑制促炎細胞因子IL-1β、TNF-α、IL-6、COX-2和組胺受體的表達有關[48]。
抗菌肽因其在宿主免疫機能中的重要地位,又被稱為宿主防御肽(Host Defense Peptides),參與機體先天性免疫反應。天然抗菌肽通常由30多個氨基酸殘基組成,是堿性小分子多肽,這些肽通常具有一定的抗炎、調控免疫活性。Epinecidin-1是從海洋石斑魚Epinepheluscoioides中分離出來的抗菌肽,能促進LPS激活的RAW264.7巨噬細胞中IL-10的分泌,但卻對TNF-α的分泌沒有明顯作用,表明該抗菌肽能通過促進促炎細胞因子的分泌來發揮抗炎作用[49]。Chrysophsin-1是從海洋真鯛Chrysophrysmajor中提取的抗菌肽,可以抑制LPS激活的RAW264.7巨噬細胞中TNF-α的釋放[50]。
3 展望
海洋生物來源的活性肽根據其化學結構的不同表現出不同的藥理作用,這些不同類型的肽對在生物醫學研究上闡明疾病機理和藥物作用機理具有重要意義。例如,環肽在過去的幾年里引起越來越多的關注,許多研究主要集中在它們在體內和體外炎癥模型中潛在的抗炎作用。對環肽的重視主要是因為其分子量小,可以保持良好的細胞通透性,同時又保證它們與藥物作用時有足夠大的接觸面積,可以擴大藥物界面,增強親和力和選擇性。因此,環肽可通過其環狀結構克服蛋白的熵(吉布斯自由能)障礙,增強與靶蛋白表面的結合。環肽的剛性是另一個關鍵因素,其可以通過顯著降低熵,更好地使目標分子與受體結合。由于這些原因,與線性結構相比,環肽通常表現出更好的生物活性。此外,由于缺乏羧基和氨基末端,環肽的環狀結構提高了它們對外肽酶水解的抵抗力。本文綜述的具有抗炎活性的海洋來源活性肽,由于環肽類物質含量低,且易降解,使得其臨床開發較難,但是隨著研究的不斷深入,以及酶工程技術和生物合成技術的成熟運用,開發海洋活性肽作為藥物先導化合物的研究有望在不久的將來得以實現。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
哲學評論(2021年2期)2021-08-22 01:53:34
學苑創造·A版(2020年9期)2020-10-13 09:41:02
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
現代企業(2015年9期)2015-02-28 18:56:50
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
土木建筑工程信息技術(2013年2期)2013-10-17 03:14:12
