線粒體介導骨骼肌重塑在運動抵御癌癥惡病質中的作用研究進展
2021-01-22 06:26:04漆正堂丁樹哲
上海體育學院學報 2021年1期
張 雪,漆正堂,丁樹哲
(1.上海體育學院附屬體育職業技術學院,上海200237;2.華東師范大學體育與健康學院,上海200241)
惡病質是一種復雜的多因素綜合征,以進行性體質量下降及肌肉減少伴或不伴脂肪減少為特點,可導致進行性多器官功能損害,其中癌癥惡病質最為常見,是20%腫瘤患者死亡的原因。現代久坐不動的生活方式破壞人們“離不開身體活動”的遺傳天性,因此,肥胖、2 型糖尿病、腫瘤等代謝性疾病相繼出現[1]。腫瘤惡病質以能量耗損、炎癥、肌量流失為主要特征。運動可能彌補了現代生活“身體活動不足”的缺陷,使能量消耗水平與遺傳基因匹配,同時阻止肌量流失,抑制癌癥惡病質發生、發展[2]。抗阻訓練、有氧訓練或相互結合作為治療癌癥的運動手段在乳腺癌[3]、肺癌[4]、前列腺癌[5]和肝癌[6]的治療中均有體現,在生理、病理、代謝、分子適應等方面也表現出積極影響。骨骼肌是運動作用的直接靶向器官,運動引起的骨骼肌一系列適應性反應通過某種直接或間接機制,抵抗癌癥惡病質發展進程。眾多研究[7-8]表明,惡病質狀態下肌萎縮與線粒體呼吸鏈活性降低、線粒體解耦連蛋白增加和線粒體功能失調有關。諸多研究[9-10]也表明,運動可通過重編程骨骼肌線粒體,修復肌纖維進而防止肌萎縮。因此,本文推測運動可通過修復惡病質狀態下的骨骼肌線粒體,重塑骨骼肌功能進而阻止癌癥惡病質發生發展,以下將對其機制進行探討。
1 癌癥惡病質骨骼肌萎縮機制
1.1 能量耗損
癌癥惡病質是典型的能量失衡癥:能量攝入減少、消耗增多,靜息代謝率提高,體質量下降。研究[11]發現:癌癥患者的骨骼肌線粒體氧化磷酸化被抑制,線粒體氧化能力下降;線粒體膜流動性改變,線粒體功能受損。
過氧化物酶體增殖物活化受體γ 輔激活蛋白?1α(peroxisome proliferator ? activated receptor gamma coactivator?1 alpha,PGC?1α)是目前已知調控線粒體生物發生最為關鍵的轉錄因子。PGC?1α 表達上調促進快肌纖維向慢肌纖維轉化,對骨骼肌細胞鈣循環進行調節;促進血管生成,提高骨骼肌的攝氧量、重塑神經肌肉接點;促進線粒體合成、脂肪合成和骨骼肌對葡萄糖的再利用,改善骨骼肌代謝[12]。腎衰竭惡病質中骨骼肌PGC?1α、線粒體轉錄因子(transcription factor A,Tfam)和線粒體融合蛋白(mitofusin?2,Mfn2)均下降,肌量下降同時伴隨骨骼肌細胞自噬增加[11,13]。在肺癌鼠模型中發現,荷瘤后1 周小鼠腓腸肌PGC?1α蛋白表達量下降40%,視萎縮蛋白1(optic atrophy type 1,Opa1)下降45%;線粒體分裂蛋白1(mitochon‐drial fission 1,Fis1)蛋白表達量在荷瘤4 周后上升80%,線粒體動態平衡被打破[11]。在惡病質狀態下骨骼肌線粒體自噬相關蛋白Bnip3 表達上調,線粒體生物發生PGC?1α 和線粒體融合蛋白Mfn2下調,線粒體分裂先于惡病質肌萎縮發生[11]。以上研究均表明,惡病質微環境中骨骼肌線粒體合成、線粒體動態平衡和線粒體自噬均發生紊亂,線粒體質量控制失衡。因此,在惡病質肌萎縮發生前重塑線粒體功能,可能是預防其發生的有效機制之一。
在惡病質狀態下,骨骼肌線粒體ATP 生成減少,能量代謝穩態失衡。研究[14]發現,在腫瘤鼠模型中,骨骼肌ATP 合成速率下降,線粒體質子電化學梯度在惡病質微環境下被打亂。在正常情況下線粒體抑制肌質網Ca2+釋放,調控鈣釋放單位的氧化還原反應。肌質網和線粒體的雙向交流為Ca2+的釋放和再攝取、ATP 的產生和再利用以及骨骼肌的生物合成提供了強有力的控制平臺。在癌癥發展期,骨骼肌鈣泵處于激活狀態,肌質網釋放大量Ca2+,ATP消耗猛增。在癌癥病理狀態下因線粒體功能受損,氧化磷酸化被抑制,ATP 生成減少,肌質網 Ca2+?ATP 酶供應不足,肌質網攝取Ca2+能力下降,導致胞內Ca2+超載;同時,從肌質網釋放到胞漿中的大量Ca2+向線粒體轉運,迫使線粒體膜通透性轉運孔(mitochondrial permeability tran‐sition pore,MPTP)過度開放,H+也隨之進入線粒體基質,加劇ATP 合成障礙[15]。肌漿中鈣超載,引起肌原纖維過度收縮,肌纖維斷裂,進而發生肌萎縮[16]。可見,減緩肌質網Ca2+釋放,避免線粒體Ca2+超載,是預防惡病質狀態下骨骼肌能量耗盡和疲勞發生的重要機制[17]。
1.2 炎癥反應
炎癥反應也是癌癥病人能量代謝異常的主要原因[18]。由感染等引發的炎癥分為可控性炎癥或急性、一過性炎癥,以及非可控性炎癥或慢性炎癥。由病毒或細菌急性感染引起短暫的一過性炎癥降低癌癥的發生率是病原相關分子模式因子(pathogen?associated molecular patterns,PAMP)激活宿主模式識別受體,包括 Toll 樣受體(toll?like receptors,TLR)、NOD 樣受體(NOD?like receptor,NLR)和RIG?Ⅰ受體(RIG?I?like receptor,RLR)等,活化先天免疫并引發抗腫瘤T細胞反應,從而引發抗腫瘤免疫保護[19]。與之相反,慢性感染引發的非可控性炎癥或慢性炎癥,動物實驗和臨床分析均表明會引發多種腫瘤,如胃癌、肝癌、乳腺癌等[18]。炎癥也是損傷相關分子模式(damage?associated molecular patterns,DAMP)激活的先天免疫反應,與肌少癥密切相關[19]。與炎癥相關的蛋白復合體感受PAMP 和 DAPM,激活Caspase?1,形成炎性體分子復合體,促進促炎因子白介素?1β(interleukin?1β,IL?1β)和白介素?18(interleukin18,IL?18)成熟及分泌,從而介導炎癥的發生[20-21]。在惡病質狀態下,骨骼肌線粒體 ROS 過量生成,誘導 NLRP3(NLR family,pyrin do‐main containing 3)和IL?1β 生成。線粒體DNA、線粒體融合蛋白、線粒體抗病毒信號通路(mitochondrial antiviral?signaling protein,MAVS)均調節 NLRP3 活性,募集炎性小體轉位到線粒體[22]。受損線粒體在胞漿中循環,加入惡病質炎癥微環境,促使肌細胞壞死。
諸多研究[23-24]表明,白介素?6(interleukin6,IL?6)在慢性疾病的炎癥發展及引起骨骼肌功能紊亂中扮演重要角色。近年來,對于IL?6 的研究[25-26]已突破原有生理病理學的認識,認為IL?6作為內分泌因子在骨骼肌代謝過程中也起到非常重要的作用。IL?6 是多效應因子,同時具有促炎和抗炎效應[27]。在多個腫瘤模型中發現,IL?6 可激活骨骼肌信號轉導、轉錄激活因子 3(signal transducer and activator of transcription 3,STAT3)和細胞外調節蛋白激酶(extracellular regu‐lated protein kinases,ERK1/2),調控骨骼肌線粒體穩態,進而調節肌細胞數量[28]。研究[29]發現,特異性敲除大鼠骨骼肌PGC?1α,骨骼肌組織和血漿中IL?6、腫瘤壞死因子α(tumor necrosis factor,TNFα)等促炎因子含量明顯增加,說明PGC?1α可抑制炎癥反應,但具體機制尚不明確。推測:PGC?1α 作為調控線粒體生物發生最為關鍵的轉錄因子,通過調節線粒體合成和線粒體融合,提升線粒體健康水平,抑制ROS?NLRP3炎性小體分泌IL?1β、IL?6 等促炎因子,從而抑制炎癥反應。研究[30]發現,腫瘤發展期骨骼肌TNF?α、IL?1β和IL?6 還可通過ATP 依賴的泛素化通路加劇蛋白水解,引起肌萎縮。此外,腫瘤微環境下的炎癥因子還可誘導一氧化氮合酶(inducible nitric oxide synthase,iNOS)表達,線粒體大量一氧化氮(nitric oxide,NO)生成也可進一步抑制氧化磷酸化,破壞骨骼肌的收縮功能[30]。可見,線粒體功能異常導致的炎癥反應在癌癥惡病質肌萎縮中發揮著重要作用。
1.3 肌量流失
癌癥惡病質機體蛋白降解增加、合成減少是惡病質最常見癥狀[31]。泛素-蛋白酶體系統(ubiquitin?proteasome system,UPS)是骨骼肌蛋白水解的重要通路,也是癌癥惡病質骨骼肌消耗的主要途徑[32]。基因測序表明,癌細胞基因組中存在多個基因發生點突變,致使蛋白質編碼序列改變多個突變蛋白表達[33]。大量突變蛋白的存在致使正常的泛素化調控蛋白降解變得異常活躍,原有的蛋白質合成機制被打破,正常蛋白質復合體的形成受影響,最終觸發泛素化介導其降解[34]。在依賴 ATP 的泛素?蛋白酶體中,E3 泛素連接酶附著于賴氨酸殘基上,通過靶向26S 蛋白酶體促進肌肉蛋白水解[35]。肌萎縮盒F蛋白?1(Atrogin?1)和肌肉特異性環指基因1(muscle?specific ring finger 1,MURF1)是E3 泛素連接酶的主要組分,激活促進底物蛋白的泛素化,促使更多蛋白轉運到蛋白酶體降解,發生肌萎縮,是癌癥惡病質骨骼肌萎縮的重要標記物[36]。胃癌病人泛素化蛋白mRNA 表達是對照組的2~3 倍,細胞內大量蛋白被UPS 降解,肌量流失,引起肌肉萎縮[37-38]。
相關研究[39-40]表明,Atrogin?1 和 MURF?1 在癌癥惡病質患者及動物模型中均表達升高,促進骨骼肌蛋白降解,促使肌發生萎縮。臨床研究[41]發現,胃癌病人的泛素化蛋白mRNA 表達是對照組的2~3 倍,體質量低于對照組8%~13%。即使在炎癥疾病中,腫瘤壞死因子?α(tumor necrosis factor,TNF?α)也可通過上調泛素載體蛋白(UbcH2/E220k)提高UPS 活性,促進骨骼肌蛋白分解代謝[42]。其原理是 TNF?α 與表面受體結合,激活 NF?kB 通路,編碼 Atrogin?1 和 MURF1基因轉錄,降解肌原纖維[43],而 NF?kB 通路又可通過E3 泛素系統保持線粒體完整性防止細胞凋亡[44]。研究[45]發現,在心肌線粒體的MURF1調節氧化反應;在對 MURF1 肌細胞定位的研究[46]中發現,轉染后的MURF1 多定位在線粒體碎片上。可見,泛素化蛋白修飾與線粒體受損密切相關。
惡病質狀態下骨骼肌蛋白除分解代謝加強外,合成代謝減少也是惡病質肌萎縮的原因。胰島素促進氨基酸向骨骼肌轉運,增加骨骼肌蛋白合成和抑制蛋白降解;胰島素敏感性降低影響骨骼肌對氨基酸的攝取,抑制蛋白質合成[47]。胰島素樣生長因子?1(insulin?like growth factor,IGF?1)是又一重要蛋白調節因子。研究[48]發現,IGF?1 所產生的多種生物學效應主要是通過激活磷脂酰肌醇3 激酶(phosphatidylinoskol 3?kinase,P13K)/Akt 信號通路發揮作用,而 Akt 激活能維持線粒體的正常功能,進而調節蛋白轉運和合成。癌癥惡病質骨骼肌IGF?1 表達下調,蛋白合成速率降低[49];轉基因鼠過表達的IGF?1 可抑制蛋白泛素化調節的肌萎縮[50],均可能與惡病質狀態下線粒體穩態被打破有關,有待進一步研究。
此外,在癌癥惡病質全身炎性反應狀態下,IL?6水平升高可誘導泛素-蛋白酶體系統引起骨骼肌蛋白降解,同時增加解偶聯蛋白表達,引起能量消耗。升高的 IL?6 參與了 UPS 的激活[51],進而誘導 E3 泛素連接酶Atrogin?1和MURF?1表達相應增加,而在線粒體膜上也存在包括 Atrogin?1 和 MURF?1 在內的多種泛素連接酶。研究[52]發現,在細胞質內加入蛋白酶體抑制劑后線粒體膜上的UCP2、UCP3 異常增加,而解偶聯蛋白UCP2、UCP3 介導的質子泄漏現象,使線粒體電子傳遞產生的質子電化學勢能被質子漏消耗,破壞膜兩側的電化學梯度,將質子電化學勢能轉化為熱量散失,引起能量消耗和代謝紊亂,促進癌癥惡病質的形成[53]。研究[54]發現,接種腹水瘤后大鼠骨骼肌中的UCP2 和UCP3 mRNA 水平明顯升高,在結腸癌、乳腺癌患者中也發現其表達上調。
以上研究表明,癌癥惡病質骨骼肌能量耗損、炎癥反應與肌量流失均與線粒體異常有關。同時,能量代謝紊亂、促炎因子的分泌和蛋白降解以線粒體為中樞形成腫瘤微環境相互影響,加快肌細胞凋亡和肌纖維損傷,加重惡病質肌萎縮發展。因此,修復惡病質狀態下線粒體結構與功能,重塑線粒體健康網絡,是抵御癌癥惡病質的關鍵,而運動很可能是修復癌癥惡病質狀態下骨骼肌異常線粒體的重要手段。
2 運動通過線粒體質量控制重塑骨骼肌
線粒體是雙層膜包裹的細胞器,在真核細胞中扮演重要角色,它是脂肪酸氧化、三羧酸循環和氧化磷酸化等重要生理生化過程發生的主要場所。線粒體處于持續動態變化,對維持細胞功能至關重要,如能量代謝和細胞凋亡等。線粒體的融合和分裂被認為是調節線粒體網絡健康程度的重要保障。處于應激狀態或受損的線粒體需被標記并選擇性地降解,以防止其與健康的線粒體整合。功能異常的線粒體會影響生物合成通路,使細胞產生過量的ROS 并激活細胞調亡通路。線粒體質量控制即線粒體生物發生、線粒體動態變化(融合與分裂)和線粒體自噬可確保線粒體數量及質量的相對穩定[55]。在腫瘤細胞中,激活的PGC?1α 引起線粒體呼吸加強,激活無效循環,促使能量消耗增加[56];但在惡病質中,骨骼肌PGC?1α、線粒體轉錄因子(transcription factor A,Tfam)和線粒體融合蛋白(mitofusin?2,Mfn2)均下降,肌量下降還伴隨著骨骼肌細胞自噬的增加[11,13]。可見,在惡病質環境中線粒體質量控制相關基因在腫瘤與骨骼肌中差異化表達。骨骼肌細胞是終末分化細胞,不具有無限增殖分化能力,不發生癌變。骨骼肌是運動作用的直接器官,運動應激下骨骼肌線粒體“重生”進而重塑骨骼肌功能可能是運動抵御惡病質肌萎縮的重要機制。修復惡病質狀態下線粒體,關鍵在于重建線粒質量控制。
2.1 PGC?1α介導骨骼肌線粒體質量控制與運動
運動可通過上調PGC?1α 重構Ca2+轉運,促使骨骼肌PGC?1α降低肌鈣蛋白表達進而誘導肌質網減少Ca2+釋放,促進快肌纖維向慢肌纖維轉化,增強骨骼肌線粒體有氧氧化能力[57]。Padrao 等[58]研究發現,35 周中等強度跑臺訓練后,乳腺癌鼠骨骼肌PGC?1α 表達上調,氧化磷酸化增強,骨骼肌ATP 合成速率增加。在癌癥惡病質發展進程中,糖酵解增強、乳酸生成增多,骨骼肌線粒體結構與功能被破壞[59]。在肥胖、糖尿病、癌癥惡病質病理發展早期階段,均發現骨骼肌乳酸生成增多,能量代謝異常[60-61]。Serge等[62]研究發現,在肌肉收縮過程中,PGC?1α 可抑制糖酵解,減少乳酸生成,抑制瓦博格效應(Warburg effect)。在運動應激條件下,細胞質中的PGC?1α 移位至細胞核內并通過與NRF1 和NRF2 共同轉錄線粒體組成蛋白及線粒體轉錄因子A(mitochondrial transcription factor A,Tfam),而Tfam 是參與線粒體DNA 復制、轉錄以及擬核形成的重要因子。另外,在運動應激下,PGC?1α 移位至線粒體并與Tfam 結合形成復合物,從而促進線粒體DNA 的轉錄和復制。由此可見,運動可通過PGC?1α 增加線粒體數量和質量,增強骨骼肌有氧氧化能力,抑制乳酸生成,重建良性的肌細胞生成環境,抵御惡病質肌萎縮。
運動還可調節線粒體自噬重塑骨骼肌功能。耐力聯合抗阻運動降低LC3B?I/II 比例,通過調節線粒體自噬,增強線粒體功能,增加骨骼肌肌量[63]。另一項比較化療與中等強度運動在抵抗癌癥惡病質肌萎縮的研究發現,早期進行運動干預可逆轉因惡病質引起的骨骼肌線粒體自噬增加,恢復線粒體功能,增加骨骼肌肌量和肌力[64]。同一課題組研究還發現,中等強度運動預防惡病質肌量流失與運動引起的低水平骨骼肌氧自由基(Reactive Oxygen Species,ROS)、羧蛋白和線粒體自噬流相關[65]。Pin 等[66]研究發現,低強度耐力運動后荷瘤組小鼠骨骼肌PGC?1α mRNA和蛋白表達均顯著上升,自噬相關蛋白表達LC3B 和Bnip3 顯著下降,受損線粒體被清除,線粒體結構和功能恢復,肌量流失減少。可見,運動可通過PGC?1α調節線粒體自噬清除損傷線粒體,修復惡病質狀態下骨骼肌受損線粒體,進而恢復肌肉功能。
2.2 IL?6、泛素化介導線粒體質量控制與運動
IL?6 是調節惡病質狀態下骨骼肌線粒體質量重塑的重要因子。研究發現,有規律的運動讓惡病質狀態下的骨骼肌,即使IL?6過表達也不會發生肌量丟失和代謝紊亂[67]。癌前期患者體質量下降,過表達的IL?6促使骨骼肌PGC?1α和線粒體融合蛋白Mfn1、Mfn2表達下調,線粒體數目未發生變化;在癌變過程中,線粒體分裂蛋白FIS1表達上調,線粒體內容物減少。在IL?6 誘導惡病質模型中發現,雖然血液循環中保持高水平的IL?6,中等強度運動可逆轉因IL?6 誘導引起的線粒體失調,抑制惡病質發展[68]。即使在非惡病質狀態下,IL?6 也可直接激活骨骼肌 DRP?1 和 FIS1 表達[69]。以上研究表明,運動并未通過降低IL?6水平抑制炎癥,但可逆轉因IL?6 引起的線粒體功能異常,重塑惡病質狀態下的骨骼肌。運動似乎可使骨骼肌對惡病質炎癥環境“脫敏”,讓骨骼肌不被其腫瘤微環境影響,進而恢復肌細胞各組件包括線粒體的正常運轉。這可能與IL?6 是多效應影響因子有關,IL?6 同時具備“促炎”和“抗炎”效應。在惡病質發生發展期,IL?6 促進炎癥反應;運動干預后,IL?6 轉變角色,抑制炎癥反應。運動如何讓IL?6“化弊為利”,具體機制有待進一步研究。
此外,研究[70]顯示,規律運動可顯著降低機體UPS 活性,抑制 MuRF1 和 MAFbx 的表達及骨骼肌蛋白降解,改善骨骼肌萎縮。有氧訓練抑制骨骼肌蛋白泛素化,逆轉因惡病質引起的肌萎縮[70-72]。抗阻運動降低骨骼肌泛素化相關基因atrogin?1 和MuRF?1 mRNA 表達,減緩肌萎縮速度[73]。低強度運動通過抑制UPS 通路,增加低氧誘導因子(hypoxia?inducible factor?1alpha,HIF?1α)和磷酸化腺苷酸活化蛋白激酶(AMP activated protein kinase,AMPK),阻止因癌癥惡病質引起的肌量流失[74]。AMPK 是一種保守的細胞和線粒體能量感受器,能磷酸化絲氨酸/蘇氨酸蛋白激酶?失調 51 樣激酶?1(uncoordinated 51 like kinase?1,ULK?1),而后者是啟動細胞自噬和線粒體自噬的關鍵調控分子。泛素-蛋白酶體系統還可通過調節線粒體蛋白質更新(turnover)和/或蛋白活性控制線粒體的蛋白質平衡,這也是線粒體質量控制的一種重要機制。UPS 受損和線粒體功能失調均與癌癥顯著相關,有效的UPS 對于維持線粒體健康至關重要。目前關于線粒體蛋白泛素化在惡病質肌萎縮中的研究較少,基于泛素化通路在維持線粒體健康中的作用以及運動抑制骨骼肌泛素化,從而阻止肌萎縮的研究基礎,將來可進行運動介導蛋白泛素化在修復惡病質狀態下線粒體功能的作用研究,進一步明確運動的抗癌機制。
總之,雖運動精準治療癌癥的具體運動效應尚未明確,美國癌癥協會仍建議癌癥病人治療期每周進行150 min 中等強度或75 min 高強度運動。癌癥惡病質患者仍可通過運動提高肌力和體能,阻止體質下降。本文總結線粒體介導運動抵御癌癥惡病質肌萎縮的作用機制見圖1。
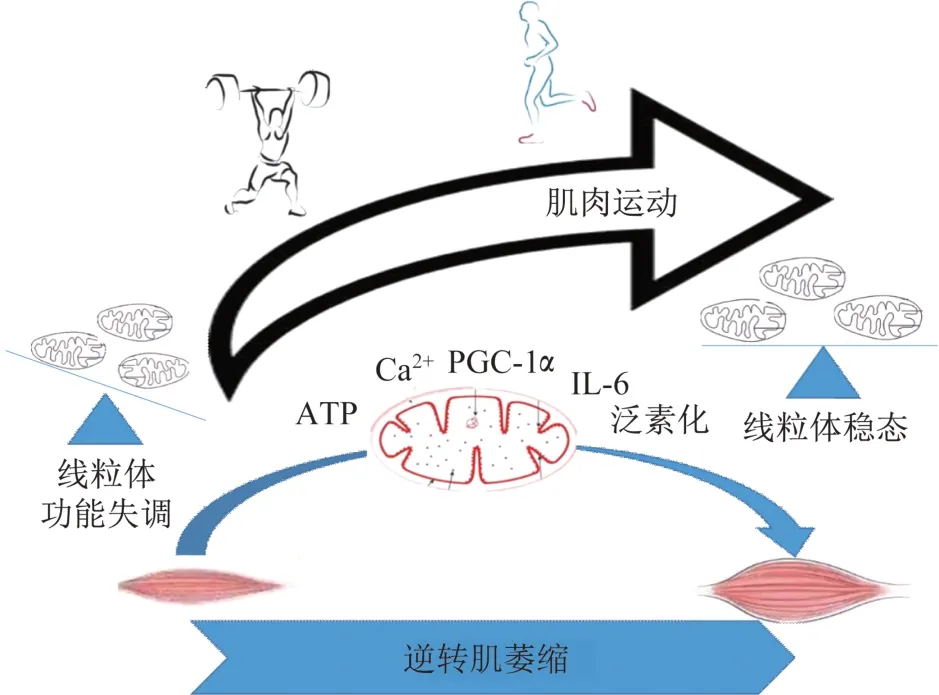
圖1 運動促進惡病質狀態下線粒體骨骼肌穩態Figure 1 Increased muscle use improves skeletal muscle mitochondrial homeostasis in cachectic conditions
3 結論與展望
近年來,骨骼肌線粒體在惡病質肌萎縮中的作用逐步成為新的研究靶點,癌癥惡病質狀態下骨骼肌線粒體功能異常引起能量代謝異常、炎癥反應和肌量流失。在惡病質微環境中,運動可通過重塑線粒體保持骨骼肌代謝穩態和逆轉肌萎縮,但仍有以下問題有待解決:①運動中生成的乳酸與腫瘤細胞的代謝產物乳酸,對骨骼肌和腫瘤的代謝意義不盡相同;②蛋白泛素化在惡病質肌萎縮發生發展中起著關鍵作用,在維持線粒體功能中也發揮作用,運動如何調控線粒體泛素化進而阻止惡病質狀態下肌萎縮發生,需進一步探明。同時,運動干預作為治療手段也面臨巨大的挑戰,在癌癥發展早期,為癌癥惡病質患者提供個性化且有效的靶向運動方案,進行基于運動反應組學分析的精準運動干預極為重要。
作者貢獻聲明:
張 雪:提出論文主題,設計論文框架,撰寫、修改論文;
漆正堂:審核、指導修改論文;
丁樹哲:提出選題思想。
