大孔樹脂純化薜荔莖總黃酮的工藝研究
2021-01-28 04:32:36王曉華韋漢燕桂勁松韋巧慧
中國民族民間醫藥 2020年24期
關鍵詞:黃酮
王曉華 韋漢燕 桂勁松 韋巧慧
桂林醫學院,廣西 桂林 541199
薜荔(FicuspumilaL.)來源于桑科榕屬[1],主要分布在廣西、湖南、四川等省區,其有相當膨大的成熟花托,像無花果,俗稱“鬼球、鬼饅頭”,最早于明朝的《本草綱目》中記載。薜荔在我國藥用歷史悠久,根、莖、葉和果實為藥用部位,具有祛風除濕、固腎填精、活血通絡、消炎解毒、抗菌、增強免疫力、抗腫瘤、抗感染、消炎鎮痛、驅蟲等作用[2]。近年研究結果顯示,薜荔植株的各部位均含有黃酮類物質,尤以莖部含量豐富。黃酮類化合物是一類有抗氧化作用的化合物,其生理活性強,能抗動脈硬化,降低膽固醇,抗痙攣、抗輻射[3]。薜荔植株中的黃酮含量頗高,具有很高的開發利用價值,但目前缺乏對薜荔莖中黃酮類成分的研究。本實驗選擇AB-8大孔吸附樹脂,通過靜態吸附和動態解吸試驗,篩選出薜荔莖總黃酮純化的最佳工藝條件,為薜荔莖總黃酮的開發利用提供參考。
1 儀器與材料
1.1 材料 薜荔莖藥材,購自廣西桂林中草藥批發市場,經桂林醫學院王曉華副教授鑒定為桑科榕屬薜荔FicuspumilaLinn的干燥莖;蘆丁對照品(中國藥品生物制品鑒定所);亞硝酸鈉(AR 汕頭市西隴化工股份有限公司,批號:130902);九水合硝酸鋁(AR 汕頭市西隴化工股份有限公司,批號:140304);氫氧化鈉(AR 武漢宏大化學試劑廠);AB-8型大孔樹脂(天津市海光化工有限公司)。
1.2 主要儀器 CP225D電子分析天平;SHZ-B水浴恒溫振蕩器(上海博迅實業有限公司醫療設備廠);UV-1600PC型紫外可見分光光度計(上海美譜達儀器有限公司);SHB-IIIT循環水式多用真空泵(鄭州長城科工貿有限公司);RE-5203旋轉蒸發儀(上海亞榮生化儀器廠)。
2 方法與結果
2.1 薜荔莖總黃酮的測定
2.1.1 對照品溶液的制備 精密稱取蘆丁對照品10.05 mg干燥至恒重,置于50 mL容量瓶中,用70%乙醇溶液溶解,稀釋至刻度,搖勻,即濃度為0.201 mg/mL溶液。
2.1.2 最大吸收波長的確定 準確吸取蘆丁對照品溶液1.0 mL,置于25 mL容量瓶中,加入5%的亞硝酸鈉溶液1.0 mL將其振蕩后靜置6 min,加入1.0 mL的10%硝酸鋁溶液,將其振蕩放置6 min后加入4.0 mL的4%氫氧化鈉溶液,最后用70%乙醇溶液調至刻度線[4],振勻放置15 min顯色。以不含對照品溶液的相應試劑作為空白對照,測定200~800 nm 掃描波長范圍內的吸光度,得到最大吸收波長為503 nm。因此,以503 nm作為測量波長。
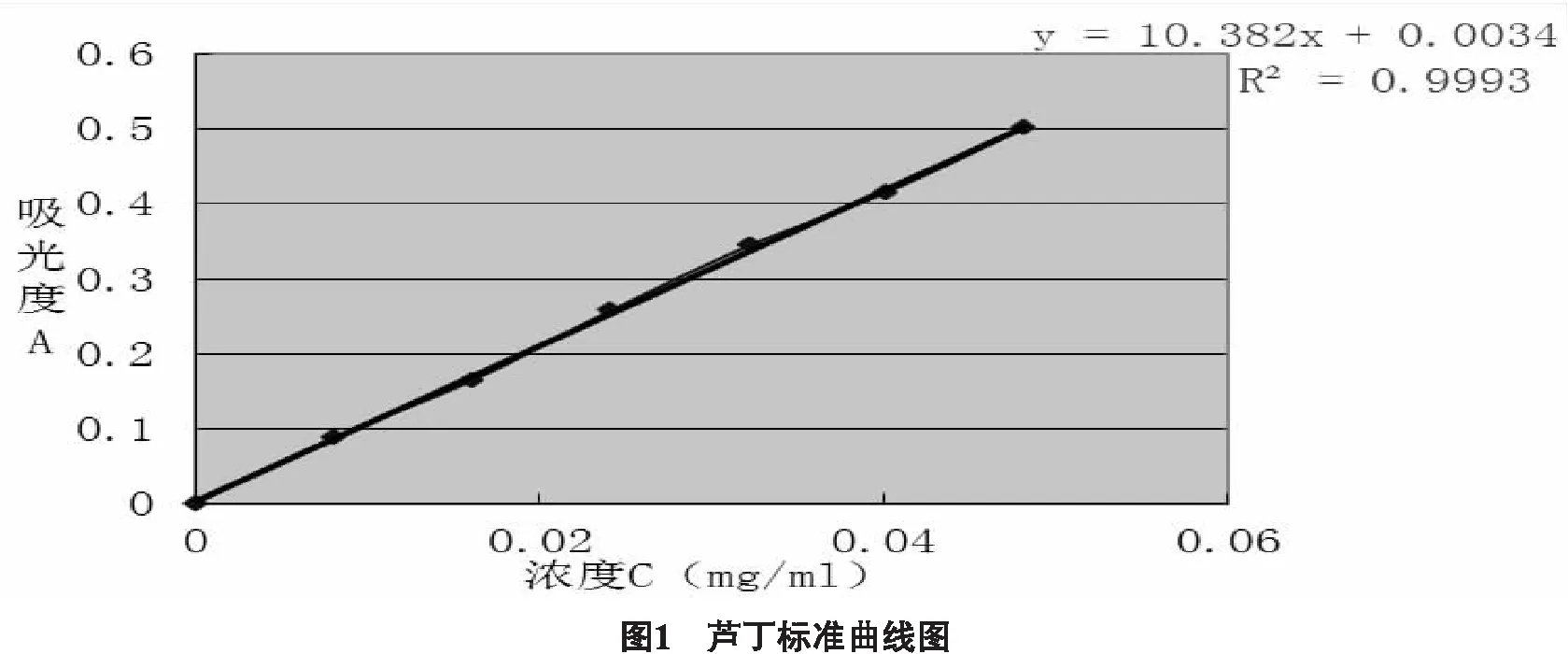
2.1.3 線性關系的確定 準確吸取蘆丁對照品溶液0、1.0、2.0、3.0、4.0、5.0、6.0 mL,置于25 mL容量瓶中,照“2.1.2”項下方法,以對照品溶液的濃度為橫坐標,吸光度為縱坐標,測定503 nm處的吸光度,得到標準曲線:A=10.382 C+0.0034(r=0.9993),結果如圖1所示。蘆丁濃度為0~0.0482 mg/mL時線性關系較好。
2.1.4 薜荔莖提取液的制備 將薜荔莖藥材粉碎,過80目篩。取100 g薜荔莖粉末,置2000 mL圓底燒瓶中,加入十倍量70%乙醇浸漬30 min,裝上冷凝回流裝置,加熱回流提取共3次,每次1 h,趁熱抽濾[5],最后用旋轉蒸發儀減壓濃縮至濃度為1 g生藥材/mL薜荔莖提取液,作為樣品溶液。
2.1.5 薜荔莖總黃酮的含量測定 精密吸取1.0 mL薜荔莖提取液,用70%乙醇稀釋至10 mL,搖勻,再從中吸取0.5 mL放置于25 mL容量瓶中。然后按“2.1.2”項下方法加入顯色試劑,以70%乙醇和相應的顯色劑為空白,測定503 nm處的吸光度值,并根據回歸方程計算總黃酮含量[6]。得薜荔莖總黃酮含量為6.25 mg/mL。
2.2 大孔樹脂的預處理 取適量的AB-8大孔樹脂置于燒杯中,用適量95%酒精浸漬AB-8大孔樹脂24 h, 使其充分膨脹后采取濕法進行裝柱,用95%酒精動態沖洗樹脂,再用蒸餾水將樹脂沖洗直到嗅不到酒精味;將5%鹽酸加到柱子里浸泡樹脂2 h后用蒸餾水動態沖洗樹脂使洗脫液的pH值呈中性, 然后將5% NaOH溶液加到柱子中浸泡樹脂2 h后用蒸餾水動態沖洗樹脂至洗脫液呈中性;用95% 乙醇沖洗樹脂使洗脫液與水混合(1∶5)至無白濁現象產生, 最后用蒸餾水沖洗樹脂至洗脫液無酒精味, 留著備用[7]。
2.3 AB-8樹脂吸附因素的考察
2.3.1 AB-8大孔吸附樹脂靜態吸附和靜態解吸實驗
2.3.1.1 靜態吸附 準確稱取經過處理的2.0 g(濕質量) AB-8大孔樹脂放入50 mL具塞三角燒瓶中,加入30 mL薜荔莖提取液(1 g生藥材/mL)。將塞子蓋緊并于37℃水浴中振蕩12 h。取上清液測定吸光度。根據標準曲線方程計算總黃酮的濃度,得出靜態吸附率和吸附容量。 吸附量Q=(C0-Cr)V/W。在公式中,Q是吸附量(mg·g-1樹脂), C0是初始質量濃度 (mg/mL),Cr是吸附后的黃酮含量 (mg/mL),V是溶液的體積 (mL), W是樹脂的質量(g)。吸附率=(吸附前黃酮含量/吸附后黃酮含量)/吸附前黃酮含量×100%。
由表1可知,樹脂靜態吸附量為50.03 mg/g,吸附率為53.36%。

表1 薜荔莖總黃酮在AB-8大孔樹脂的吸附量
2.3.1.2 靜態解吸 將已靜態吸附的飽和大孔樹脂用蒸餾水沖洗到洗脫液呈無色。然后加入70%乙醇30 mL并在37℃水浴振蕩器上搖動12 h解吸。解吸后,通過吸取上清液測定吸光度。根據標準曲線方程計算總黃酮的濃度,得出靜態解吸率。解吸率D(%)=(V×Cr)/(W×D)×%。式中:Q為吸附量(mg·g-樹脂);Cr為解吸后溶液中黃酮的含量(mg/mL);V是溶液的體積(mL);W為樹脂的質量(g)。由表2可知,靜態解吸后的解吸率為84.61%。

表2 薜荔莖總黃酮的洗脫率
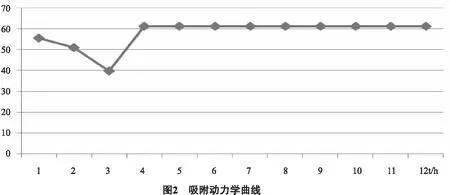
2.3.1.3 靜態吸附動力學研究 稱量2.0 g(濕質量)已處理過的樹脂置于50 mL的帶塞三角瓶中,再加入30 mL濃度為1 g生藥材/mL的薜荔莖提取液,在恒溫器 37℃處晃動2 h, 每1 h吸收一次上清液, 并測定吸光度值。以標準曲線方程確定總黃酮濃度及吸附率, 將時間作為橫向坐標, 吸附率為縱向坐標,繪制動力學曲線。從圖2結果可以看出, AB-8大孔樹脂對薜荔莖總黃酮的吸附是一種快速平衡型吸附,隨著吸附時間的增加,吸附速率在3 h內迅速下降,4 h后吸附速率穩定,吸附平衡基本達到。因此,吸附時間被確定為4 h。薜荔莖總黃酮的富集純化使用AB-8大孔樹脂適宜。
2.3.2 吸附條件的考察
2.3.2.1 樣品液的質量濃度的影響 分別加入70%的乙醇溶液稀釋已制得的薜荔莖1 g生藥材/mL濃度的提取液,得到質量濃度分別為0.25、0.50、0.75 g生藥材/mL的樣品液。稱量4份已預處理過的 AB-8大孔樹脂,每2.0 g(濕質量)為一份于50 mL的具塞三角瓶中,分別加入質量濃度為0.25、0.50、0.75 g生藥材/mL的樣品液30 mL,在恒溫水浴振動篩上于37℃放置4 h,然后取上清液測量吸光度,得出總黃酮的濃度與吸附率。由表3可以看出,當該樣品液濃度為0.5 g生藥材/mL時,薜荔莖總黃酮的吸附率最佳,因此選擇0.5 g生藥材/mL作為樣品液的質量濃度。
2.3.2.2 pH值的影響 稱取5份已預處理的AB-8大孔樹脂,每2.0 g(濕質量)為1份,把它們分別放進一個50 mL的錐形瓶里,將濃度為1 g生藥材/mL的提取液30 mL加入瓶中, pH調節至3、4、5、6、7在37℃下振蕩3 h。測量吸光度,計算得出總黃酮的濃度與吸附率。從表4可以看出,當pH在3 ~ 6范圍時,AB-8大孔樹脂對薜荔莖總黃酮具有較好的吸附率,當吸附介質pH值為7時,吸附率明顯提高, 因此,吸附劑的pH值調到7最優。

表3 不同的提取液濃度對AB-8大孔樹脂吸附容量的影響

表4 PH值對AB-8大孔樹脂的吸附量影響
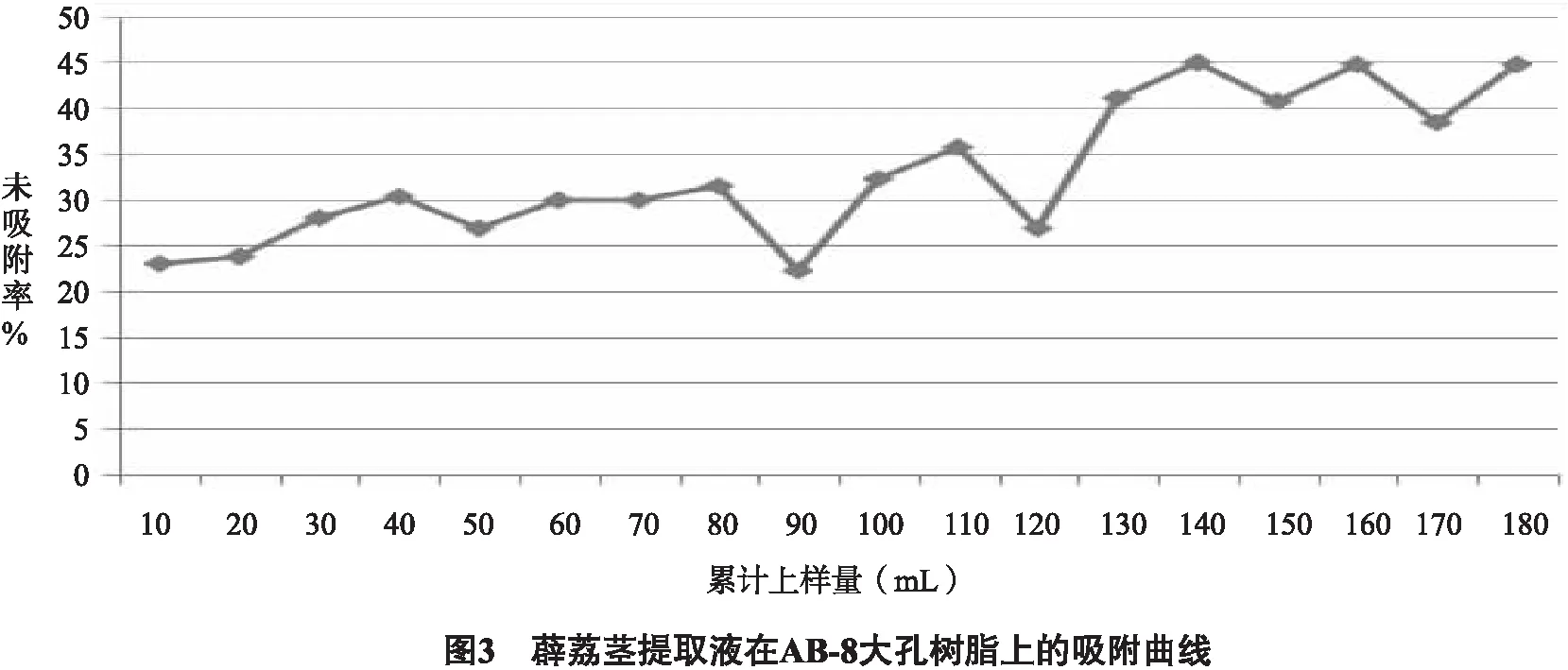
2.3.2.3 泄漏曲線的繪制與上樣量的確定 準確稱取5.0 g經過預處理的AB-8大孔樹脂,濕法裝柱,取0.5 g生藥材/mL濃度(pH調為7)的薜荔莖提取液,樣品流速為1 mL/min, 每10 mL收集1個樣品, 收集18份樣品,測定吸光度值并計算總黃酮的濃度, 將藥物溶液體積作為橫坐標,將未吸附率繪制在縱坐標上以確定樣品上柱體積。未吸附率=流出液中成分含量/上柱前原液中流出物含量。圖3結果顯示,上樣量達140 mL時發生明顯泄漏,樹脂柱不能完全吸附藥液中的黃酮成分,所以上樣量為140 mL時更合理,故確定140 mL(14 BV)作為最大上樣量。
2.3.3 洗脫條件的考察
2.3.3.1 洗脫劑體積分數的影響 取5份已預處理的AB-8大孔樹脂,分別放到50 mL的具塞三角瓶中,并加入0.5 g生藥材/mL(pH=7)薜荔莖提取液適量使其吸附達到飽和,再加入不同體積分數的洗脫劑30 mL(50%、60%、70%、80%、90%乙醇),放置于37℃的恒溫水浴中于振蕩器搖動4 h,取其上清液,以吸光度值來計算解吸速率。從表5可以看出,當乙醇的體積分數小于70%時,解吸首先下降然后上升,然后緩慢上升。因此,選取的洗脫劑為70%乙醇。

表5 不同乙醇濃度的影響
2.3.3.2 確定洗脫劑的用量 準確稱量5.0 g的AB-8大孔樹脂,濕法裝柱。取140 mL濃度為0.5 g生藥材/mL(pH調至7)的薜荔莖提取液,流速為1 mL/min上樣。用5倍柱體積(5BV)的純水沖洗樹脂除去雜質,用70%乙醇以1.0 mL/min的流速洗脫至洗脫液呈無色,收集10 mL/份的洗脫液來測定吸光度。用于計算總黃酮含量。以總洗脫量為縱坐標 (mg) ,洗脫量 (mL)為橫坐標,得到動態洗脫曲線。 總洗脫量(mg)=Cd2×Vd2。式中,流出物的總黃酮質量濃度為Cd2,洗脫劑體積為Vd2。從圖4可以看出,當洗脫液為80 mL(約8BV)時,莖中的總黃酮已被完全洗脫,因此選擇8BV作為洗脫劑的量。
2.4 工藝驗證試驗 對上述所得到的最優工藝條件進行3次重復試驗驗證,計算大孔樹脂富集純化后薜荔中總黃酮的質量分數。經過3次驗證試驗,從表6可知大孔樹脂富集純化后的薜荔莖總黃酮的質量分數分別為65.04%、63.56%、64.37%,平均質量分數是64.32%,其結果表明,AB-8大孔樹脂對薜荔莖總黃酮的富集純化過程是穩定可行的。
根據上述實驗的結果,確定了薜荔莖總黃酮的純化工藝條件是:取薜荔莖提取液(0.5 g生藥材/mL),藥液 pH=7,上樣量14 BV,以1 mL/mL的速度通過 AB-8大孔吸附樹脂柱,靜置4 h,再用5 BV的蒸餾水來沖洗水溶性雜質,用8 BV 70%乙醇以1 mL/min的速度洗脫大孔樹脂,將洗脫液減壓濃縮。回收溶劑,烘干并干燥,得樣品。
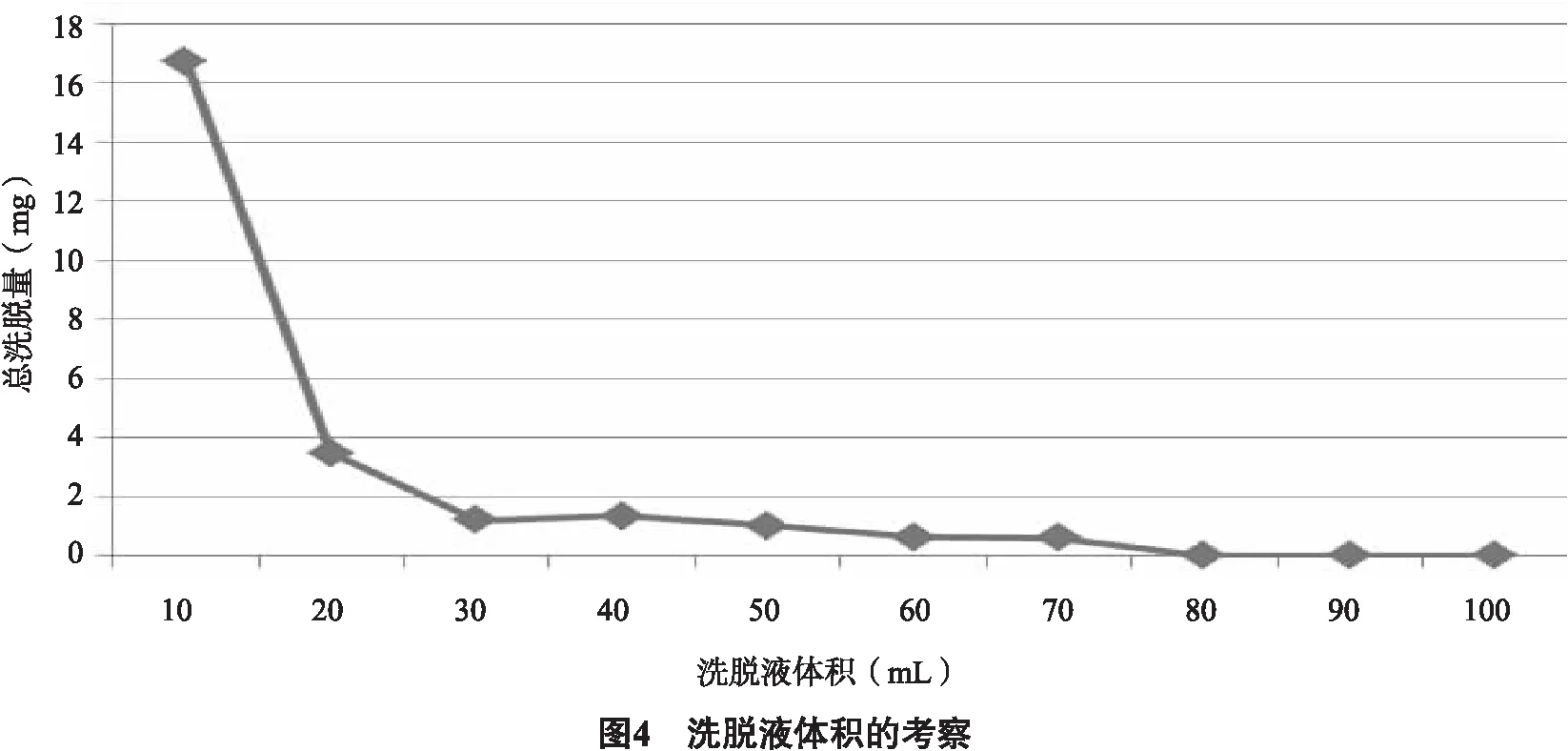

表6 薜荔莖總黃酮的質量分數
3 討論
AB-8型大孔吸附樹脂是一種具有極大比表面積、合適孔徑的球形弱極性聚合物吸附樹脂,它可通過表面吸附、氫鍵等作用于黃酮類化合物,是黃酮類化合物的優良吸附劑。本實驗從靜態吸附和動態解吸兩個方面對AB-8大孔吸附樹脂分離純化薜荔莖總黃酮的工藝條件進行了研究。結果表明: 樣品液的濃度、pH值、樣品體積、靜態吸附時間、乙醇體積分數、洗脫劑用量對薜荔莖總黃酮的吸附量和洗脫率影響較大。當乙醇的濃度小于70%時,解吸率呈先下降后上升再緩慢增長趨勢。從實驗結果可以看出,可選擇70% ~ 90% 的乙醇體積分數來洗脫。由于考慮到實驗室的經濟效益方面,本實驗以70%乙醇作為洗脫液濃度。實驗結果顯示,50.03 mg/g為AB-8型大孔吸附樹脂對薜荔莖總黃酮靜態的吸附量,而84.61%是解吸附率,呈現出其具備較好的分離純化能力。最優的吸附工藝條件是:取薜荔莖提取液(0.5 g生藥材/mL),藥液 pH=7,上樣量14 BV,以1 mL/mL的速度通過 AB-8大孔吸附樹脂柱,靜置4 h,再用5 BV蒸餾水來沖洗水溶性雜質,用8 BV 70%乙醇以1 mL/min的速度洗脫大孔樹脂,其洗脫液用旋轉蒸發儀進行減壓濃縮回收乙醇,干燥后得到紅褐色膏狀物。該工藝簡單易操作,無污染,樹脂可再生,生產效率高,具有一定的推廣應用價值。
猜你喜歡
四川蠶業(2021年2期)2021-03-09 03:15:32
四川蠶業(2021年3期)2021-02-12 02:38:46
中成藥(2018年11期)2018-11-24 02:57:00
中成藥(2017年8期)2017-11-22 03:19:40
中成藥(2017年10期)2017-11-16 00:50:13
中成藥(2017年4期)2017-05-17 06:09:50
哈爾濱醫藥(2016年1期)2017-01-15 13:43:16
天然產物研究與開發(2016年11期)2016-06-15 20:29:17
湖南師范大學自然科學學報(2015年1期)2015-02-27 14:50:04
安徽醫藥(2014年12期)2014-03-20 13:15:15
