超高效液相色譜三重四極桿質(zhì)譜聯(lián)用測定食品湯汁中的毒品
2021-02-10 07:14:00羅達(dá)龍
食品安全導(dǎo)刊 2021年35期
王 華,羅達(dá)龍,覃 藍(lán)
(梧州市食品藥品檢驗(yàn)所,廣西梧州 543002)
苯丙胺(AMP)、甲基苯丙胺(MA)、3,4-亞甲基二氧基甲基苯丙胺(MDMA)、3,4-亞甲二氧基-N-乙基-苯丙胺(MDEA)和3,4-亞甲基二氧基苯丙胺(MDA)為具有苯丙胺結(jié)構(gòu)的物質(zhì),該類化學(xué)組分具有中樞興奮的作用,服用后會產(chǎn)生致幻、精神依賴性、食欲不振和擬交感能效應(yīng)等藥理與毒理學(xué)特征。在我國,該類物質(zhì)是受管制的精神活性物質(zhì),故濫用潛力較大[1]。氟胺酮(2F-DCK)為氯胺酮苯環(huán)上的氯元素被氟元素所取代,在我國氟胺酮不屬于管制類藥物,在公安偵辦的涉毒案中也屢屢查獲該物質(zhì)[2-4]。王世玉等[5]于1987年報道了氟胺酮的合成方法,并證明了化合物氟胺酮具有與氯胺酮類似的麻醉作用。氟胺酮能被歸為麻醉活性物質(zhì)的一種,也存在被濫用的潛力。
為保障人們舌尖上的安全,有必要建立一種操作簡單、定性/定量準(zhǔn)確、測試靈敏度高的檢測方法。對于上述物質(zhì)目前可用的方法有快檢法、高效液相色譜法(High Performance Liquid Chromatography,HPLC)和動態(tài)液相微萃取(Gas Chromatography/Mass Spectrometry in the Selected Ion Monitoring Mode,GC/MS-SIM)等方法。由于火鍋湯中存在的基質(zhì)種類較多,如凈化不完全會使雜質(zhì)帶入系統(tǒng),產(chǎn)生基質(zhì)干擾,影響試驗(yàn)回收率,甚至?xí)饳z驗(yàn)結(jié)論判斷錯誤和儀器耗損嚴(yán)重。
本研究將乙腈作為提取溶液,使用Captiva EMRLipid固相萃取柱進(jìn)行凈化,并結(jié)合UPLC-MS/MS、ACE C18-苯基色譜柱,通過多反應(yīng)監(jiān)測(Multiple Reaction Monitoring,MRM)對火鍋湯汁中的AMP、MA、MDMA、MDEA、MDA和2F-DCK進(jìn)行檢測。
1 材料與方法
1.1 儀器與試劑
安捷倫超高效液相色譜儀1290-三重四極桿液質(zhì)聯(lián)用儀6470(美國安捷倫公司),超純水器(賽多利斯Arium Comfort Ⅱ);超聲儀;高速冷凍離心機(jī);Captiva EMR-Lipid固相萃取柱(1 mL,40 mg,安捷倫科技有限公司)。
乙腈(質(zhì)譜級,歐姆尼公司);檢驗(yàn)用水為經(jīng)賽多利斯Arium Comfort Ⅱ純水機(jī)制備的去離子水;苯丙胺類混合標(biāo)準(zhǔn)品購自于安捷倫科技有限公司,質(zhì)量濃度100 μg/mL;氟胺酮標(biāo)準(zhǔn)品質(zhì)量濃度100 μg/mL,購買于阿爾塔公司。
1.2 色譜條件
分析用色譜柱:ACE C18-苯基柱(2.1 mm×100 mm,2 μm);流動相:0.2%甲酸-15 mmol/L乙酸銨(A)-乙腈(B);流速:0.35 mL/min;進(jìn)樣體積:2 μL;柱溫箱溫度:35 ;液相色譜流動相洗脫程序:0~ 1.50 min,10% ~ 20% B;1.50~ 3.50 min,20%~35% B;3.51~4.50 min,95% B,后運(yùn)行時間:1.00 min。
1.3 質(zhì)譜條件
ESI離子源正模式采集,干燥氣體溫度與干燥氣體流量:150 ,12 L/min;鞘氣加熱溫度與鞘氣流量:350 ,12 L/min;噴霧器壓力:25 Psi;毛細(xì)傳輸管電壓:3 500 V;化合物離子對、碰撞能量AMP:136→ 119(5 V)91(17 V);MA:150→ 91(17 V)119(5 V);MDMA:194→ 163(9 V)105(25 V);MDEA:208→105(25 V)163(9 V);MDA:180→163(5 V) 135(18 V);2F-DCK:222→163(20 V) 109(40 V)。
1.4 標(biāo)準(zhǔn)溶液的制備
精密吸取苯丙胺類標(biāo)準(zhǔn)品和氟胺酮標(biāo)準(zhǔn)品各1 mL,置于10 mL經(jīng)標(biāo)定合格的容量瓶中,用乙腈溶劑稀釋標(biāo)品溶液至刻度,得到濃度為10 μg/mL的對照品保存液。用乙腈將對照品保存液進(jìn)行梯度稀釋,得到濃度分別為S1:0.02 ng/mL、S2:0.1 ng/mL、S3:0.2 ng/mL、S4:1.0 ng/mL和 S5:2.0 ng/mL的標(biāo)準(zhǔn)溶液。
1.5 供試樣品的制備
取火鍋湯樣品,在9 000 r/min,4 條件下離心4 min,吸取離心后的上層清湯液體5.00 mL至25.00 mL容量瓶,用乙腈溶劑定容至刻度,搖勻。取稀釋液約3 mL,在15 000 r/min,4 條件下離心3 min。吸取經(jīng)離心后的上清液1 mL過EMR凈化柱,收集后用有機(jī)濾膜過濾,上機(jī)測定。
2 結(jié)果與分析
2.1 線性關(guān)系考察
取本文“1.4”節(jié)中配制的S1~S5,按“1.2~1.3”條件,上儀器進(jìn)行測定,通過儀器采集軟件采集各物質(zhì)的峰面積。以配制的標(biāo)準(zhǔn)曲線濃度(x,ng/mL)為橫坐標(biāo),采集所得的峰面積(y)為縱坐標(biāo),使用儀器自帶工作站進(jìn)行線性回歸,各物質(zhì)線性回歸方程如下。AMP:y=34 524.298 821x-755.830 859,r=0.999 2;MA:y=53 280.726 060x-986.025 191,r=0.999 6;MDMA:y=55 132.373 168x-1 236.319 062,r=0.999 7;MDEA:y=34 724.761 291x-761.259 699,r=0.999 2;MDA:y=22 939.079 908x-538.973 499,r=0.999 2;2F-DCK:y=4 1025.108 859x-765.748 774,r=0.999 6,各物質(zhì)在線性濃度范圍0.02~2.00 ng/mL時,物質(zhì)線性的相關(guān)系數(shù)均不小于0.995 0。
2.2 回收率試驗(yàn)
將標(biāo)準(zhǔn)溶液加入至空白火鍋湯樣品中,采用本文“1.5”節(jié)方法進(jìn)行處理,測定AMP、MA、MDMA、MDEA、MDA和2F-DCK 6個物質(zhì)的回收率,6個物質(zhì)在0.1~5.0 ng/mL的加標(biāo)水平下,樣品的回收率為83.2%~94.0%,RSD為3.1%~4.6%,見表1。
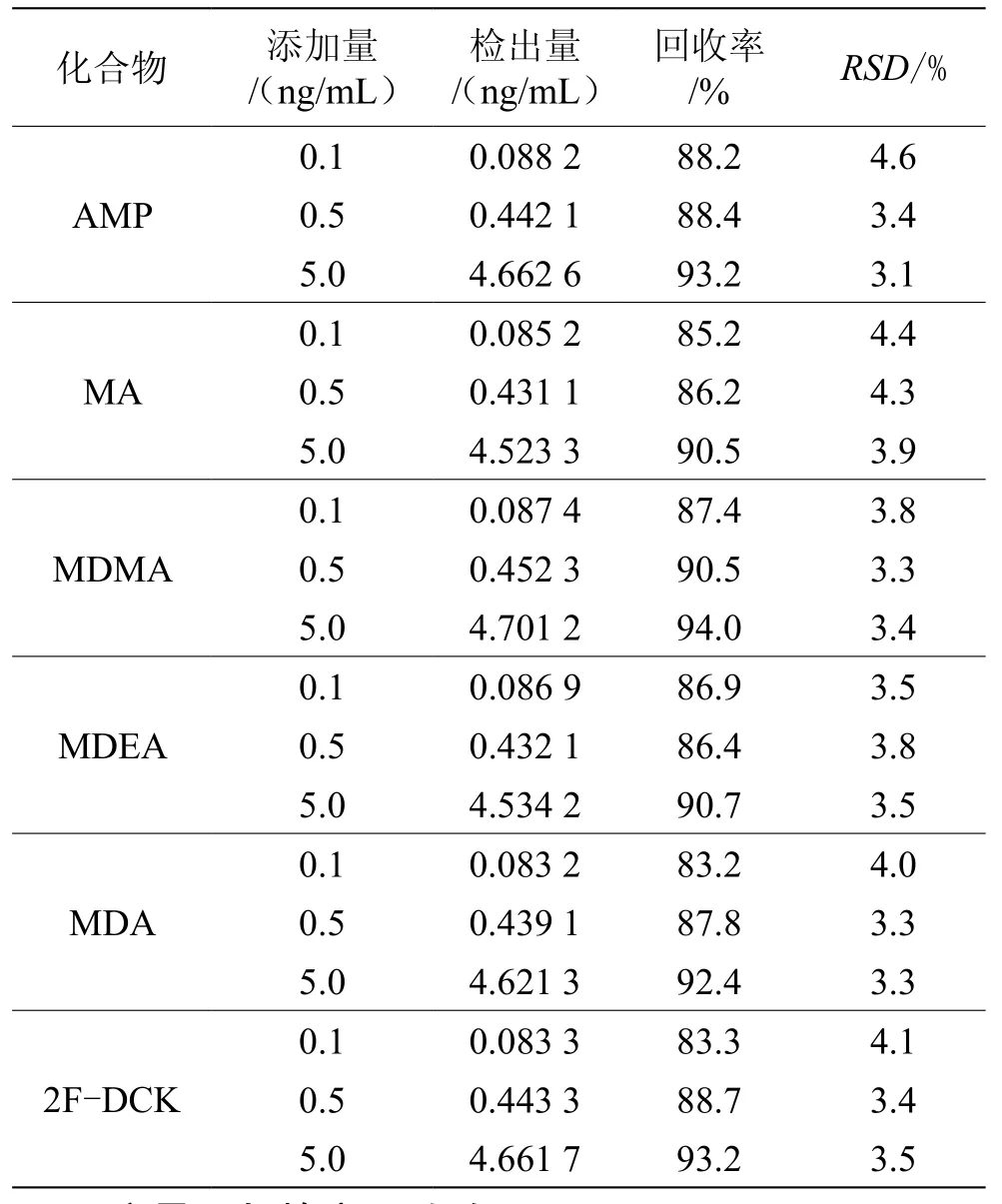
表1 基質(zhì)加標(biāo)回收率結(jié)果(n=3)
2.3 定量限與檢出限試驗(yàn)
取本文“1.4”節(jié)下的標(biāo)準(zhǔn)溶液,進(jìn)行級別稀釋,按“1.2色譜條件、1.3質(zhì)譜條件”進(jìn)樣測定。當(dāng)工作站計算被測定物質(zhì)的信噪比不小于10∶1時,即為定量限;當(dāng)工作站計算被測定物質(zhì)的信噪比不小于3∶1時,即得出檢出限。由表2可知,6個物質(zhì)的定量限均為0.1 ng/mL,檢出限均為0.03 ng/mL。
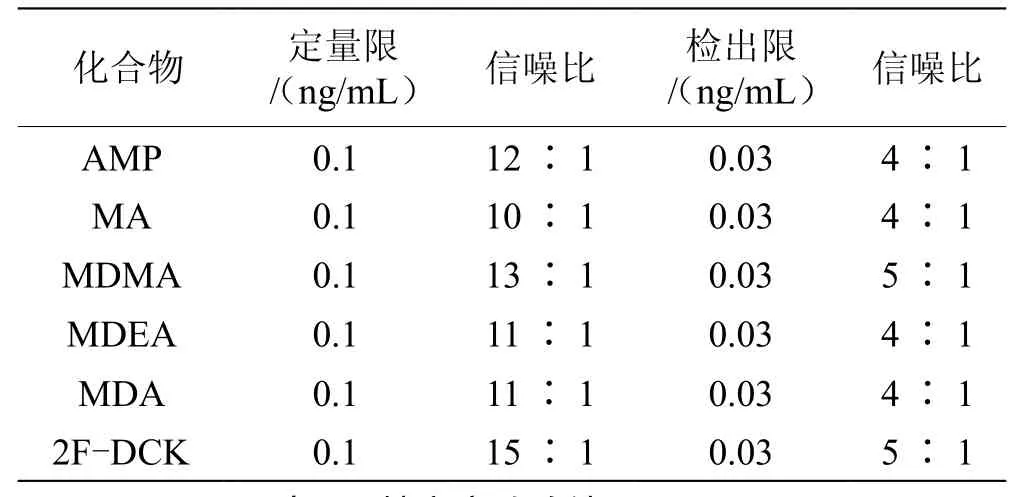
表2 定量限與檢出限試驗(yàn)結(jié)果
2.4 儀器精密度試驗(yàn)
取本文“1.4”節(jié)下的標(biāo)準(zhǔn)溶液S3,按“1.2~1.3”條件連續(xù)進(jìn)樣檢測6次,各物質(zhì)峰面積的RSD見表3。AMP:RSD=1.5%;MA:RSD=1.6%;MDMA:RSD=1.4%;MDEA:RSD=1.1%;MDA:RSD=1.6%;2F-DCK:RSD=1.6%,表明儀器精密度良好。
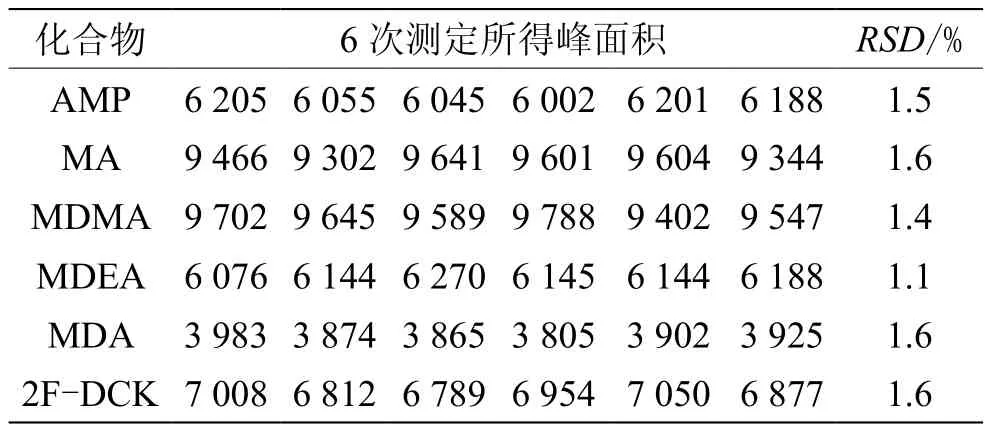
表3 精密度試驗(yàn)結(jié)果(n=6)
2.5 色譜柱與流動相的考察
方法考察中選擇水與乙腈體系、0.2%甲酸水溶劑與乙腈混合體系、0.2%甲酸-15 mmol/L乙酸銨與乙腈溶劑混合體系等組成進(jìn)行流動相適用性考察,分別在C18色譜柱與C18-苯基柱進(jìn)行試驗(yàn)。結(jié)果發(fā)現(xiàn)在0.2%甲酸-15 mmol/L乙酸銨-乙腈作為流動相時,能有效促進(jìn)6個化合物的電離,6個化合物的峰面積達(dá)到最大,且得到更優(yōu)的靈敏度。而在C18-苯基柱上測定6個化合物得到的色譜峰峰形優(yōu)于C18色譜柱。因此,選擇0.2%甲酸-15 mmol/L乙酸銨-乙腈作為本次試驗(yàn)的流動相,選用C18-苯基柱作為本試驗(yàn)的色譜柱。6個化合物的色譜圖見圖1。
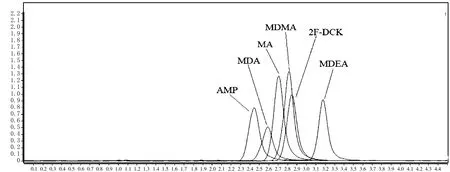
圖1 化合物色譜圖
2.6 樣品測定
購買市售的火鍋湯汁共15份按“1.2~1.5”條件進(jìn)行前處理并測定,結(jié)果15份樣品中均未檢出AMP、MA、MDMA、MDEA、MDA 和 2F-DCK 6種物質(zhì)。
3 結(jié)論
本研究建立了運(yùn)用Captiva EMR-Lipid固相萃取柱對火鍋湯中的基質(zhì)進(jìn)行凈化前處理,通過使用液質(zhì)聯(lián)用儀的多反應(yīng)監(jiān)測模式,結(jié)合C18-苯基色譜柱對苯丙胺、甲基苯丙胺、3,4-亞甲基二氧基甲基苯丙胺和3,4-亞甲二氧基-N-乙基-苯丙胺、3,4-亞甲基二氧基苯丙胺和氟胺酮進(jìn)行定性定量檢測。從被測樣品的前處理到檢驗(yàn)完成整個試驗(yàn)耗時約30 min。本方法具有操作簡單、快速、定性與定量準(zhǔn)確及靈敏度高等特點(diǎn),能用于日常檢驗(yàn)。本方法使用的固相萃取柱具有高選擇性,能有效去除樣品基質(zhì)中的脂質(zhì),所含技術(shù)為體積排阻和疏水性相互作用的原理對脂質(zhì)進(jìn)行凈化。脂質(zhì)的去除能減少對質(zhì)譜電噴霧離子化的影響,使待測組分在分析過程中不受干擾,以進(jìn)一步提高檢測方法的穩(wěn)定性與數(shù)據(jù)的可靠性,減少了儀器的維護(hù)成本。
