SO2和O3對錳-鈷催化劑氧化NO的影響研究
2021-02-21 16:05:10絲夢沈伯雄
河北工業大學學報 2021年6期
絲夢 沈伯雄
摘要 采用浸漬法制備了15%Mn-Co(2∶1)/TiO2催化劑,研究其催化氧化NO的性能,重點研究了SO2和O3對NO催化氧化性能的影響。性能測試和傅里葉變換原位紅外光譜分析結果表明,SO2在催化劑表面以物理和化學吸附作用為主,生成的硫酸鹽物種占據了催化劑活性位點,抑制了催化劑對NO的吸附和催化氧化。O3能夠促使硫酸鹽物種向覆蓋力較弱的吸附態轉變,有利于釋放催化劑表面活性位點。此外,O3能夠明顯促進單配位硝酸鹽的生成,特別是在低溫條件下,有利于硝酸鹽物種釋放出氣態NO2。因此,O3能夠提高催化劑抗SO2的性能,從而提高其對NO的催化氧化能力。
關 鍵 詞 NO氧化;臭氧;抗硫性能;錳-鈷催化劑;原位紅外光譜;機理分析
中圖分類號 X701.3;O643.36? ? ?文獻標志碼 A
文章編號:1007-2373(2021)06-0069-08
Abstract 15%Mn-Co(2∶1)/TiO2 catalyst was prepared by impregnation method to study the catalytic oxidation of NO, with emphasis on the influences of SO2 and O3 on the performance of NO catalytic oxidation by the catalyst.The results of activity experiments and in situ FTIR showed that, the physical and chemical adsorption of SO2 on the catalyst surface was dominant, and the generated sulfate species occupied the active sites, which inhibited the adsorption and catalytic oxidation of NO by the catalyst. However, the presence of O3 can promote the transformation of sulfate species to a weak adsorption state, which was conducive to the release of active sites on the catalyst surface. In addition, O3 can significantly promote the formation of monodentate nitrates, especially at low temperature, which was conducive to the release of gaseous NO2 by nitrate species. Therefore, O3 can improve the tolerance of 15%Mn-Co(2∶1)/TiO2 catalyst to SO2,thus improving its catalytic oxidation performance of NO.
Key words NO oxidation; ozone; sulfur resistance; manganese-cobalt catalyst; in situ FTIR; mechanism analysis
0 引言
隨著工業技術的發展,化石燃料燃燒造成的空氣污染已經成為不可回避的話題。電力、鋼鐵、冶金、焦炭等行業以及我國北方冬季供暖的煤炭、天然氣燃燒均會產生大量氮氧化物(NOx)、SO2、粉塵顆粒以及揮發性有機物等空氣污染物。對我國京津冀地區在2017—2018年供暖季PM2.5主要組分占比進行統計,硝酸鹽已取代硫酸鹽成為細顆粒污染物中最主要的二次無機組分[1]。這意味著相較以往強調SO2的排放控制,NOx的排放控制越發具有緊迫性和重要性。在過去的二十年里,人們對控制NOx排放技術的研究以選擇性催化還原(SCR)和選擇性非催化還原(SNCR)技術為主[2-4]。但反應溫度要求較高、還原劑NH3的逃逸等問題依然存在。并且煙氣中的SO2會與還原劑NH3反應生成(NH4)2SO4和(NH4)HSO4,造成設備腐蝕、煙氣管道堵塞及催化劑減活等[5]。
近年來,氧化法脫除煙氣中的NOx備受關注[6-9]。高價態的氮氧化物溶解度更高,并且氧化法能夠同時氧化煙氣中的單質汞和有機物等,后續與濕法脫硫相結合,在一體化同時脫除煙氣中多污染物的應用中具有較高潛力。O3作為一種強氧化劑,且分解產物為O2,沒有二次污染,因此O3氧化煙氣中的NO得到一定的研究和應用[10-11]。但煙氣中存在的SO2在O3聯合催化劑氧化NO的過程中,會明顯抑制NO的氧化[12],而對其抑制機理目前還不明晰,有必要開展深入研究。本研究制備了負載型15%Mn-Co(2∶1)/TiO2催化劑,結合原位紅外光譜分析討論了高溫下(350 ℃)和低溫下(100 ℃)SO2和O3對Mn-Co氧化物催化劑催化氧化NO的影響進行了研究。
1 實驗方法
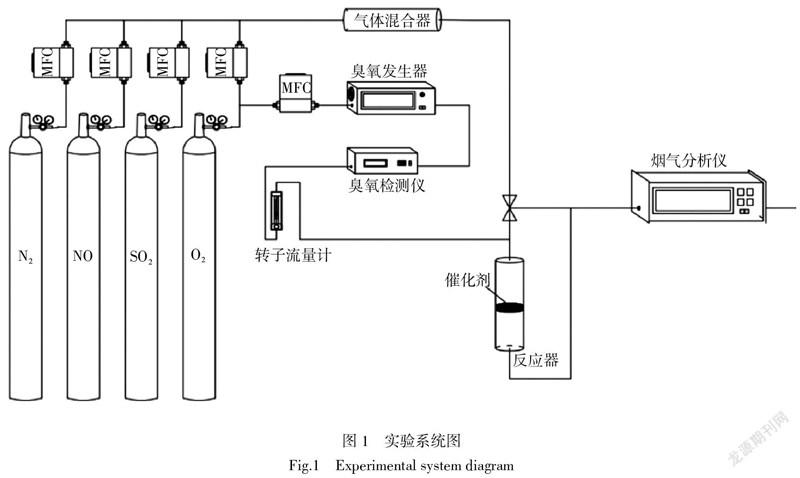
以銳鈦型TiO2為載體,浸漬法制備了總負載量為15 %(質量分數)的錳、鈷氧化物催化劑,其中錳、鈷元素物質的量比為2∶1,500 ℃空氣氣氛下煅燒4 h,記為15%Mn-Co(2∶1)/TiO2催化劑,研磨成40~60目顆粒用于催化氧化NO的實驗。實驗系統圖如圖1所示,O3由氧氣源臭氧發生器產生,其濃度經臭氧檢測儀檢測。模擬煙氣由N2作為載氣,總流量為400 mL/min,NO初始濃度為500×10-6,SO2初始濃度為300×10-6,O2含量為4%,O3初始濃度為250×10-6,體積空速GHSV=24 000 h-1。煙氣分析儀測量反應器進出口的NO濃度和SO2濃度,以便得到NO的氧化效率和SO2的反應效率。傅里葉變換原位紅外光譜用TENSOR II(Bruker)紅外分析儀采集。分辨率為4 cm-1,掃描次數為32次。在測試前催化劑樣品經N2在400 ℃下吹掃1 h,去除表面吸附性雜質后,分別在100 ℃和350 ℃下收集背景信號,然后通入反應氣體,采集所需紅外圖譜。在原位紅外測試中,煙氣各組分濃度保持與臺架實驗中各氣體濃度一致,總流量為40 mL/min,GHSV=24 000 h-1。
2 實驗結果
2.1 SO2在催化劑表面的吸附性能
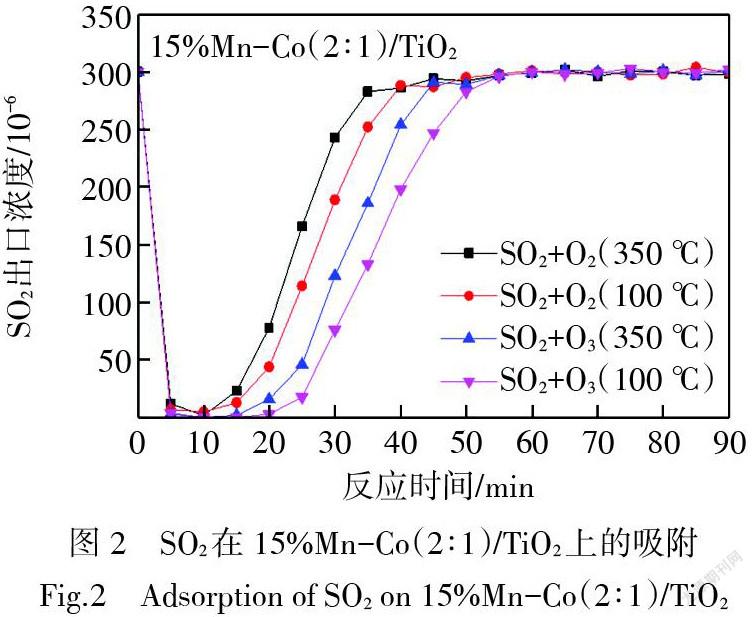
圖2為SO2與O2或O3持續同時通過15%Mn-Co(2∶1)/TiO2催化劑表面后SO2濃度的變化情況。可以看到在氣體通過催化劑的前幾分鐘反應器出口的SO2濃度迅速降至接近0,之后SO2出口濃度逐漸升高;隨著時間延長,反應器出口SO2濃度接近其初始濃度,說明SO2與O2或O3在15%Mn-Co(2∶1)/TiO2催化劑上主要發生的是吸附反應,當吸附飽和后,SO2出口濃度上升到入口濃度的水平。在體積空速為24 000 h-1的條件下,300×10-6 SO2與4% O2在350 ℃下同時吸附,約35 min SO2達到吸附飽和;在100 ℃下同時吸附,約40 min SO2達到吸附飽和;300×10-6 SO2與250×10-6O3在350 ℃下同時吸附,在45 min左右SO2達到吸附飽和;在100 ℃下同時吸附,接近50 min SO2達到吸附飽和。SO2低溫吸附飽和時長高于高溫吸附飽和時長。與SO2和O2的同時吸附相比,O3的引入能夠延長SO2在催化劑15%Mn-Co(2∶1)/TiO2上達到吸附飽和的時間,這可能是由于O3對催化劑具有一定的活化能力,增加了催化劑上對SO2吸附的活性位點數量,從而提高了催化劑對SO2的吸附量。
2.2 SO2對NO催化氧化效率的影響
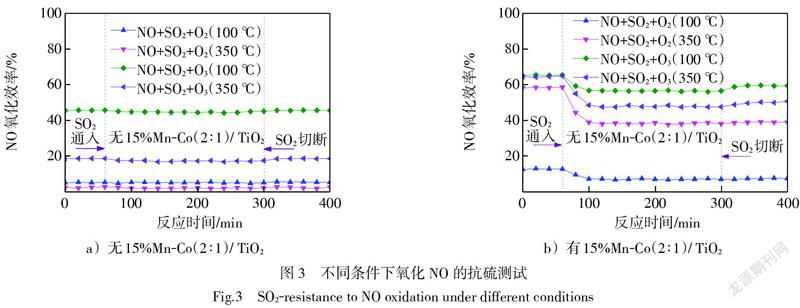
為考察SO2對NO催化氧化效率的影響,分別在100 ℃和350 ℃下對O3或O2均相氧化NO,以及O3或O2聯合15%Mn-Co(2∶1)/TiO2催化氧化NO的性能進行了研究,結果如圖3所示。圖3a)表明,在不使用催化劑的條件下,煙氣中含有O3或者O2,NO的均相氧化在高溫和低溫下受SO2的影響均不明顯。這一現象在Wang等[13]對O3氧化法同時脫硫脫硝的研究中同樣得到印證。這是因為在均相氧化中SO2與O2或O3反應所需的活化能遠高于NO氧化所需的活化能[14],因此在氧化競爭反應中NO更占優勢,SO2幾乎不參與反應。在100 ℃下O2氧化NO的效率高于350 ℃下的,是因為O2均相氧化NO的反應是放熱反應,溫度升高不利于反應發生[15]。由于O3在高溫下大量分解[12],因此在100 ℃下O3氧化NO的效率顯著高于350 ℃下的。圖3b)表明,在先前的研究中[12],15%Mn-Co(2∶1)/TiO2催化劑在350 ℃時活性最高,因此在350 ℃下催化O2氧化NO的效率高于100 ℃下的。并且,催化劑的使用提高了NO的氧化效率,特別是在沒有SO2的情況下,在100 ℃和350 ℃下催化O3氧化NO的效率幾乎一樣。然而,SO2的引入致使NO的氧化效率明顯降低,這與已報道的錳-鈷氧化物催化劑用于催化O2氧化NO的研究結果一致[16]。但O3的存在能夠削弱SO2對NO催化氧化的不利影響,并且在停止通入SO2后,NO的催化氧化效率有輕微恢復,說明O3的加入能夠改善15%Mn-Co(2∶1)/TiO2催化氧化NO的抗硫能力。
3 傅里葉變換原位紅外光譜分析
3.1 SO2在催化劑表面的吸附
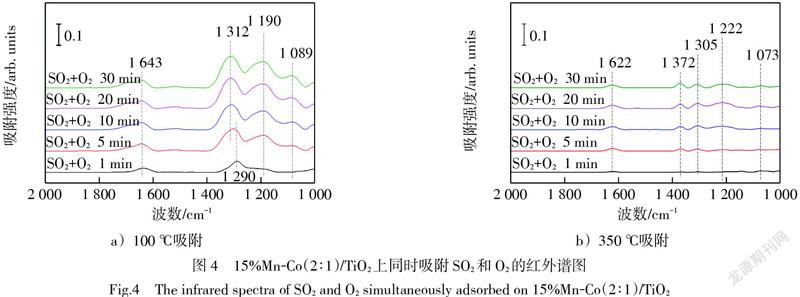
為探究O3提高SO2在催化劑表面吸附能力的原因,利用原位紅外光譜儀研究了SO2和O2以及SO2和O3在15%Mn-Co(2∶1)/TiO2催化劑表面吸附的過程。如圖4a)所示,在100 ℃下SO2與O2同時吸附,在1 643 cm-1和1 290 cm-1處最先出現了硫酸鹽物種的特征峰。其中,1 643 cm-1處的特征峰可以歸屬為催化劑表面殘余羥基與SO2反應所致[17-18],1 290 cm-1處的特征峰可以歸屬為催化劑表面形成的塊狀硫酸鹽的伸縮振動所致[19]。隨著吸附時間推移,特征峰1 190 cm-1和1 089 cm-1逐漸顯現,歸屬為表面硫酸鹽物種[18],而特征峰1 290 cm-1偏移至1 312 cm-1的位置。有研究認為這3個特征峰代表了雙配位硫酸鹽的三分裂ν3峰[20-21]。典型的雙配位雙核硫酸鹽的三分裂ν3峰譜帶位于1 050 cm-1至1 250 cm-1的范圍,若二氧化硫吸附形成的是雙配位單核硫酸鹽,則其三分裂ν3峰的譜帶會向高波數方向移動[22]。因此,這里將1 312 cm-1、1 190 cm-1和1 089 cm-1處的硫酸鹽物種歸為雙配位單核硫酸鹽的特征峰。隨吸附時間延長,硫酸鹽物種逐漸累積,各特征峰逐漸增強。圖4b)中SO2和O2在350 ℃下吸附,除了出現位于1 622 cm-1、1 305 cm-1、1 222 cm-1和1 073 cm-1處的前述硫酸鹽物種的特征峰,還在1 372 cm-1的位置出現了共價基團O=S=O的非對稱性振動特征吸附峰,該特征峰是以SO32-形態弱吸附在催化劑上的[23],在高溫下易被分解,釋放出氣態SO2,也有研究將該特征峰認為是氣態SO2的特征峰[14, 24]。這解釋了圖2中高溫下SO2的吸附飽和時間較低溫下更短的現象。
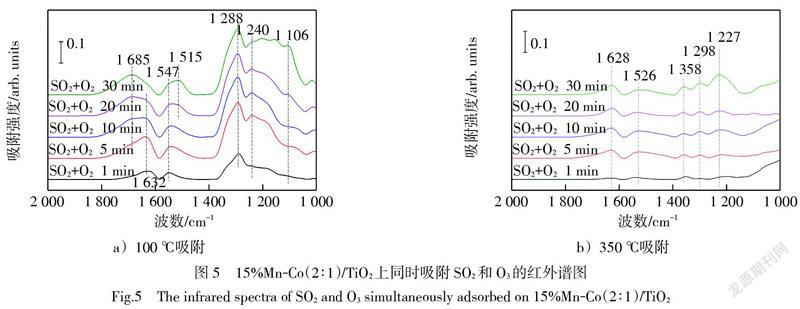
圖5a)中在100 ℃下SO2和O3的同時吸附出現了強烈的催化劑表面羥基與SO2反應的特征峰(1 632 cm-1和1 547 cm-1),并隨吸附時間延長逐漸增加,移動至1 685 cm-1和1 515 cm-1的位置,意味著O3的存在強化了催化劑上表面羥基與SO2的反應。在1 288 cm-1、1 240 cm-1和1 106 cm-1處形成的雙配位單核硫酸鹽的三分裂ν3的特征峰,隨吸附時間顯著增強,說明SO2與O3在催化劑表面形成了大量的硫酸鹽,特別是1 240 cm-1和1 106 cm-1的增長速度快于1 288 cm-1。圖5b)中350 ℃下SO2和O3的同時吸附同樣出現了氣態SO2的特征峰(1 358 cm-1)[24],雙配位單核硫酸鹽在1 227 cm-1處特征峰的增長相較于其他幾個特征峰更為明顯。但相較于圖5a)中,歸屬于雙配位單核硫酸鹽三分裂ν3峰的低波數特征峰在1 200 cm-1至1 050 cm-1之間難以觀察到。
圖4中SO2和O2同時吸附時,1 312 cm-1至1 290 cm-1之間的特征峰增長相對明顯;而圖5中SO2與O3同時吸附時,1 240 cm-1至1 106 cm-1之間的特征峰增長相對明顯。說明SO2與O3在催化劑表面吸附更傾向于形成低波數的雙配位硫酸鹽,而SO2與O2在催化劑表面吸附更傾向于形成高波數的雙配位硫酸鹽。而高波數的硫酸鹽被認為是聚集在催化劑表面的塊狀硫酸鹽,對催化劑活性位點覆蓋作用更強,而低波數的表面硫酸鹽吸附狀態相對較弱,對催化劑活性位點覆蓋作用相對較弱[19]。這也反應了O3能夠提高催化劑對SO2吸附量的原因。
3.2 SO2對NO在催化劑表面吸附的影響
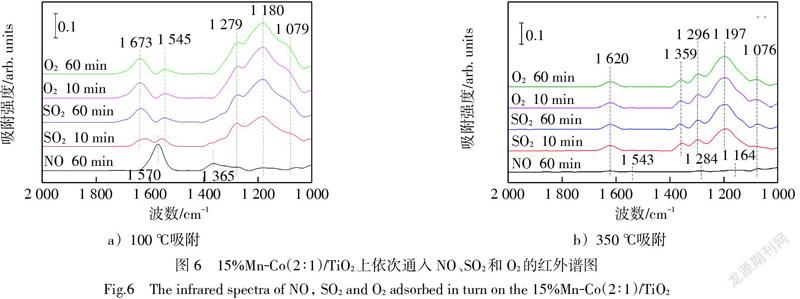
為探究SO2對催化氧化NO的影響機制,在原位紅外反應池中依次通入NO、SO2和O2(O3),各1 h,記錄紅外吸收峰的變化情況。如圖6a),100 ℃下NO吸附在新鮮15%Mn-Co(2∶1)/TiO2上出現了2個明顯的特征峰和3個微弱的特征峰,分別歸屬為雙配位硝酸鹽(1 570 cm-1)[25]、硝酸根離子(1 365 cm-1)[24]、單配位硝酸鹽(1 279 cm-1)[25]、橋接硝酸鹽(1 180 cm-1)和亞硝酸鹽(1 079 cm-1)[26]。切斷NO后通入SO2,1 570 cm-1位置處的雙配位硝酸鹽消失,取而代之的是代表SO2與催化劑表面殘余羥基形成的硫酸鹽特征峰(1 637 cm-1和1 545 cm-1)。其他硝酸鹽物種的特征峰被雙配位單核硫酸鹽的三分裂ν3峰(1 279 cm-1、1 180 cm-1和1 079 cm-1)覆蓋,因此難以分辨。停止通入SO2而引入O2后,各特征峰強度沒有明顯變化。類似地,圖6b)在350 ℃下NO吸附在新鮮15%Mn-Co(2∶1)/TiO2催化劑表面形成了5個非常微弱的硝酸鹽特征峰,分別是1 620 cm-1處的橋接硝酸鹽[25-27],1 543 cm-1處的雙配位硝酸鹽[28],1 284 cm-1處的單配位硝酸鹽[29],1 164 cm-1處的橋接硝酸鹽和1 076 cm-1處的亞硝酸鹽[26]。在通入SO2后也均被覆蓋,切斷SO2,通入O2之后,各特征峰沒有顯著變化。
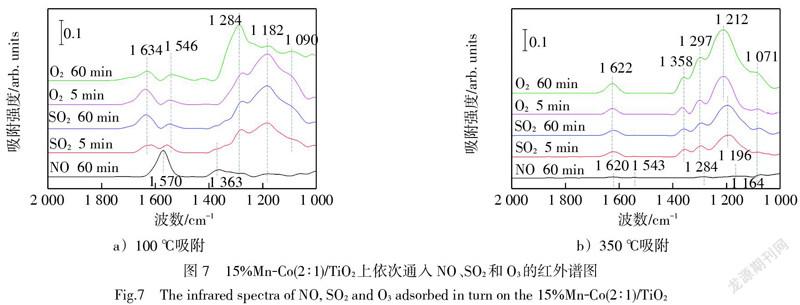
而在100 ℃下新鮮15%Mn-Co(2∶1)/TiO2催化劑上先后吸附了NO和SO2后,再通入O3,如圖7a)所示,雙配位單核硫酸鹽的三分裂ν3峰的1 182 cm-1和1 090 cm-1處的特征峰減弱,而1 284 cm-1處的特征峰增強,說明O3與催化劑上吸附的表面硫酸鹽物種發生了反應。但特征峰被覆蓋的硝酸鹽物種也可能與O3發生反應使1 284 cm-1處的特征峰增強,累積更多的單配位硝酸鹽。類似地,在圖7b)中350 ℃下新鮮15%Mn-Co(2∶1)/TiO2催化劑上先后吸附了NO和SO2后,硝酸鹽物種的特征峰被硫酸鹽物種的特征峰覆蓋,再通入O3,所有特征峰均增強。對比圖6和圖7,可以發現O3相較于O2,更易促進各吸附物種的增加和減少,說明O3更有利于反應的進行。
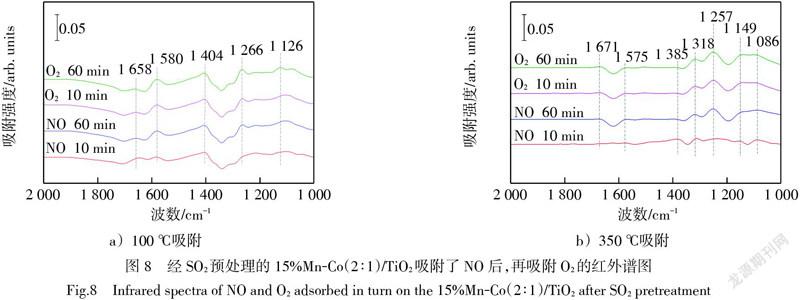
然而,由于后吸附的SO2在催化劑表面形成了大量硫酸鹽物種,覆蓋了先前吸附的硝酸鹽物種,因此難以觀察到O2及O3引入后硝酸鹽物種的變化情況。為進一步說明硝酸鹽物種受SO2的影響,以及O3的促進作用,交換NO與SO2的吸附先后順序,在經300×10-6 SO2預吸附1 h后的15%Mn-Co(2∶1)/TiO2催化劑上,進行NO和O2或O3吸附的原位紅外實驗。如圖8所示,經SO2預處理后的催化劑再由氮氣吹掃30 min,以去除弱吸附物質的影響,并以此時預處理后的催化劑表面紅外吸收譜線為背景峰,收集NO通入后的紅外吸收峰以及O2通入后的紅外吸收峰。圖8a)在100 ℃下NO吸附到經SO2預處理的催化劑上后,形成了N2O4(1 658 cm-1)[28]、雙配位硝酸鹽(1 580 cm-1)[28]、硝酸根離子(1 404 cm-1)[30]、單配位硝酸鹽(1 266 cm-1)[25]和亞硝酸鹽(1 126 cm-1)[25] 5個特征峰。在O2通入之后,這5個特征峰基乎沒有發生變化,說明低溫下O2較難與硝酸鹽物種發生反應。
圖8b)在350 ℃下NO吸附到經SO2預處理的催化劑上,形成了N2O4(1 671 cm-1)[28]、雙配位硝酸鹽(1 575 cm-1)[25]、硝酸根離子(1 385 cm-1)[25]、1 318 cm-1(硝基化合物)[31]、單配位硝酸鹽(1 257 cm-1)[25]、橋接硝酸鹽(1 149 cm-1)和亞硝酸鹽(1 086 cm-1)[26] 7個微弱的特征峰。通入O2后,各硝酸鹽物種的吸收峰強度有輕微增加,還可以看出1 149 cm-1處的吸收峰強度最終幾乎與1 086 cm-1處的吸收峰強度持平。說明高溫下由于催化劑的活性增強,O2能夠與吸附的硝酸鹽物種發生不太劇烈的反應。這與圖3b)中SO2存在且無O3時,350 ℃下NO催化氧化效率高于100 ℃下NO催化氧化效率相對應。
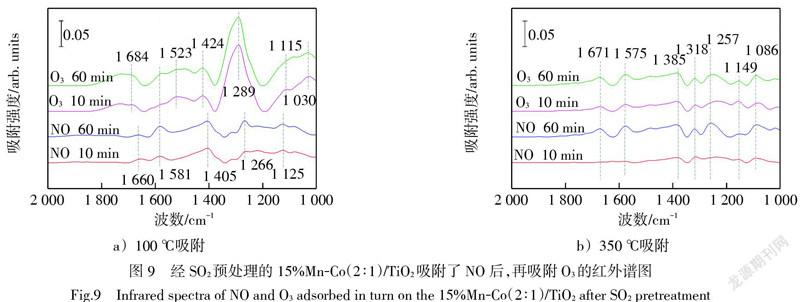
圖9為15%Mn-Co(2∶1)/TiO2催化劑經300×10-6的SO2預吸附1 h后,再用氮氣吹掃30 min,以此時的吸附狀態為背景峰,之后再吸附1 h NO,最后通入O3的紅外圖譜。從圖8和圖9中可以看出,NO吸附在經SO2預處理后的催化劑上形成的硝酸鹽物種的特征峰,其強度明顯弱于圖6和圖7中NO在新鮮催化劑上形成的硝酸鹽特征峰。這是因為SO2在催化劑表面吸附形成的硫酸鹽物種覆蓋了催化劑的活性位點,使得催化劑上能夠吸附NO的活性位點大幅減少,進而降低了能夠與O3發生反應的硝酸鹽物種的含量,使得催化氧化NO的效率降低。
圖9a)中100 ℃下經SO2預處理的催化劑吸附NO后,出現了1 660 cm-1、1 581 cm-1、1 405 cm-1、1 266 cm-1和1 125 cm-1 5個特征峰,與圖8a)中硝酸鹽物種的歸屬一致。之后隨著O3吸附,特征峰1 660 cm-1向1 724 cm-1的位置移動,1 581 cm-1處的特征峰逐漸被新出現的1 523 cm-1處特征峰覆蓋,該特征峰可以歸屬為單配位硝酸鹽[29]。1 405 cm-1移動至1 424 cm-1,1 266 cm-1移動至1 289 cm-1,1 125 cm-1移動至1 115 cm-1,并且在1 030 cm-1處出現了歸屬于反對稱N=O的特征峰[25]。可以看到在O3通入之后,特征峰1 289 cm-1的紅外吸收強度明顯增加,此特征峰歸屬于單配位硝酸鹽,并且在1 523 cm-1處也出現了單配位硝酸鹽的特征峰。單配位硝酸鹽被認為是硝酸鹽物種向氣態NO2轉化的關鍵中間產物,大量增加的單配位硝酸鹽有利于提高NO向NO2轉化的效率[12]。對比圖9a)與圖8a),在預吸附SO2的15%Mn-Co(2∶1)/TiO2催化劑表面,O3的通入能夠促使單配位硝酸鹽特征峰的明顯增長,而O2沒有此促進作用。因而O3的加入能夠增強硝酸鹽物種向氣態NO2轉變的過程,有助于提高NO催化氧化過程中對SO2的耐受能力。這解釋了圖3b)中有SO2存在時,15%Mn-Co(2∶1)/TiO2催化O3氧化NO的效率高于其催化O2氧化NO的效率的原因。
圖9b)中在350 ℃下經SO2預處理的15%Mn-Co(2∶1)/TiO2催化劑上吸附NO,形成的7個微弱的硝酸鹽特征峰,1 671 cm-1、1 575 cm-1、1 385 cm-1、1 318 cm-1、1 257 cm-1、1 149 cm-1和1 086 cm-1,其歸屬物質與圖8b)的一致。通入O3后,1 257 cm-1處雙配位硝酸鹽的特征峰有輕微波動,1 149 cm-1處橋接硝酸鹽的特征峰強度有小幅增長,其余硝酸鹽物種的特征峰沒有明顯變化。這是因為高溫下O3的分解作用加劇,削弱了其對硝酸鹽物種的氧化作用,但仍能使部分硝酸鹽物種發生轉化。這與圖3b)中催化O3氧化NO的效率在350 ℃下低于在100 ℃下的實驗結果相對應。
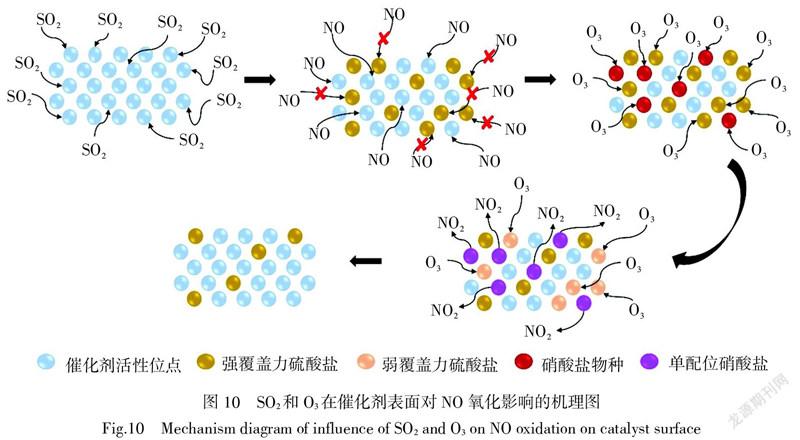
根據上述分析,SO2和O3對NO的催化氧化影響機制可以用圖10描述。SO2吸附在催化劑上覆蓋了表面活性位點,抑制了催化劑對NO的吸附從而導致NO催化氧化效率降低。O3的引入使催化劑表面覆蓋力較強的硫酸鹽物種轉化為覆蓋力較弱的硫酸鹽物種,從而逐漸釋放出活性位點用于NO的催化氧化。并且,O3易與吸附在催化劑表面的硝酸鹽物種反應,促進單配位硝酸鹽的形成,利于NO的氧化,從而提高了催化劑對SO2的耐受性。
4 結論
論文研究了浸漬法制備的負載型錳-鈷氧化物催化劑用于催化氧化NO,重點研究了SO2和O3對NO催化氧化效率的影響。SO2在15%Mn-Co(2∶1)/TiO2催化劑表面主要發生的是吸附反應,生成的硫酸鹽物種占據了活性位點,抑制了催化劑對NO的吸附和催化氧化。SO2與O2和O3的均相反應很弱,對NO的氧化沒有顯著影響。O3的引入能夠提高催化劑對SO2的吸附量及催化氧化NO過程中的抗硫性能。傅里葉變換原位紅外光譜的研究表明,相較于O2,O3更易與吸附在催化劑表面的硫酸鹽物種發生反應,特別是在低溫條件下,使催化劑表面覆蓋力較強的硫酸鹽物種向覆蓋力較弱的吸附態轉變,有利于釋放出催化劑表面的活性位點,從而提高了催化劑對NO的吸附能力。此外,低溫下O3的存在能夠顯著增強硝酸鹽物種之間的轉化,特別是促進了單配位硝酸鹽的形成,即便是在經SO2預處理后的催化劑表面,這種促進作用也是顯著的。O3的存在改善了硫中毒后的催化劑對NO的催化作用,提高了催化劑的抗硫性能。
參考文獻:
[1]? ? 彭穎蓓,劉國彬,田霄,等. 典型城市PM2. 5的污染特征、來源及影響因素分析[C]//《環境工程》2019年全國學術年會論文集,2019:171-174.
[2]? ? YANG J,SU Z H,REN S,et al. Low-temperature SCR of NO with NH3 over biomass char supported highly dispersed MnCe mixed oxides[J]. Journal of the Energy Institute,2019,92(4):883-891.
[3]? ? MLADENOVI? M,PAPRIKA M,MARINKOVI? A. Denitrification techniques for biomass combustion[J]. Renewable and Sustainable Energy Reviews,2018,82:3350-3364.
[4]? ? CHEN C M,CAO Y,LIU S T,et al. Review on the latest developments in modified vanadium-titanium-based SCR catalysts[J]. Chinese Journal of Catalysis,2018,39(8):1347-1365.
[5]? ? HU W S,ZHANG Y H,LIU S J,et al. Improvement in activity and alkali resistance of a novel V-Ce(SO4)2/Ti catalyst for selective catalytic reduction of NO with NH3[J]. Applied Catalysis B:Environmental,2017,206:449-460.
[6]? ? J?GI I,STAMATE E,IRIMIEA C,et al. Comparison of direct and indirect plasma oxidation of NO combined with oxidation by catalyst[J]. Fuel,2015,144:137-144.
[7]? ? MA Q,WANG Z H,LIN F W,et al. Characteristics of O3 oxidation for simultaneous desulfurization and denitration with limestone–gypsum wet scrubbing:application in a carbon black drying kiln furnace[J]. Energy & Fuels,2016,30(3):2302-2308.
[8]? ? WU Q,SUN C L,WANG H Q,et al. The role and mechanism of triethanolamine in simultaneous absorption of NOx and SO2 by magnesia slurry combined with ozone gas-phase oxidation[J]. Chemical Engineering Journal,2018,341:157-163.
[9]? ? LI H T,LIU F,WANG S H,et al. Oxidation and absorption of SO2 and NOx by MgO/Na2S2O8 solution at the presence of Cl-[J]. Fuel Processing Technology,2019,194:106125.
[10]? 張明慧,馬強,徐超群,等. 臭氧氧化結合濕法噴淋對玻璃窯爐煙氣同時脫硫脫硝實驗研究[J]. 燃料化學學報,2015,43(1):88-93.
[11]? ZHANG J,ZHANG R,CHEN X,et al. Simultaneous removal of NO and SO2 from flue gas by ozone oxidation and NaOH absorption[J]. Industrial & Engineering Chemistry Research,2014,53(15):6450-6456.
[12]? SI M,SHEN B X,ZHANG H H,et al. Comparative study of NO oxidation under a low O3/NO molar ratio using 15% Mn/TiO2,15% co/TiO2,and 15% Mn-Co(2:1)/TiO2 catalysts[J]. Industrial & Engineering Chemistry Research,2020,59(4):1467-1476.
[13]? WANG Z H,ZHOU J H,ZHU Y Q,et al. Simultaneous removal of NOx,SO2 and Hg in nitrogen flow in a narrow reactor by ozone injection:Experimental results[J]. Fuel Processing Technology,2007,88(8):817-823.
[14]? SUN C L,ZHAO N,ZHUANG Z K,et al. Mechanisms and reaction pathways for simultaneous oxidation of NOx and SO2 by ozone determined by in situ IR measurements[J]. Journal of Hazardous Materials,2014,274:376-383.
[15]? LIN F W,WANG Z H,ZHANG Z M,et al. Flue gas treatment with ozone oxidation:an overview on NOx,organic pollutants,and mercury[J]. Chemical Engineering Journal,2020,382:123030.
[16]? LI H H,WANG S K,WANG X,et al. Catalytic oxidation of Hg0 in flue gas over Ce modified TiO2 supported Co-Mn catalysts:Characterization,the effect of gas composition and co-benefit of NO conversion[J]. Fuel,2017,202:470-482.
[17]? ZHANG J Y,LI C T,ZHAO L K,et al. A Sol-gel Ti-Al-Ce-nanoparticle catalyst for simultaneous removal of NO and Hg0 from simulated flue gas[J]. Chemical Engineering Journal,2017,313:1535-1547.
[18]? XU W Q,HE H,YU Y B. Deactivation of a Ce/TiO2 catalyst by SO2 in the selective catalytic reduction of NO by NH3[J]. The Journal of Physical Chemistry C,2009,113(11):4426-4432.
[19]? YE D,QU R Y,SONG H,et al. New insights into the various decomposition and reactivity behaviors of NH4HSO4 with NO on V2O5/TiO2 catalyst surfaces[J]. Chemical Engineering Journal,2016,283:846-854.
[20]? ZHANG R D,ALAMDARI H,KALIAGUINE S. SO2 poisoning of LaFe0. 8Cu0. 2O3 perovskite prepared by reactive grinding during NO reduction by C3H6[J]. Applied Catalysis A:General,2008,340(1):140-151.
[21]? JIANG B Q,WU Z B,LIU Y,et al. DRIFT study of the SO2 effect on low-temperature SCR reaction over Fe-Mn/TiO2[J]. The Journal of Physical Chemistry C,2010,114(11):4961-4965.
[22]? PEAK D,FORD R G,SPARKS D L. An in situ ATR-FTIR investigation of sulfate bonding mechanisms on goethite[J]. Journal of Colloid and Interface Science,1999,218(1):289-299.
[23]? YOUN S,SONG I,LEE H,et al. Effect of pore structure of TiO2 on the SO2 poisoning over V2O5/TiO2 catalysts for selective catalytic reduction of NOx with NH3[J]. Catalysis Today,2018,303:19-24.
[24]? YU Y K,WANG J X,CHEN J S,et al. Promotive effect of SO2 on the activity of a deactivated commercial selective catalytic reduction catalyst:an in situ DRIFT study[J]. Industrial & Engineering Chemistry Research,2014,53(42):16229-16234.
[25]? HADJIIVANOV K I. Identification of neutral and charged NxOy surface species by IR spectroscopy[J]. Catalysis Reviews,2000,42(1/2):71-144.
[26]? QI G,LI W. NO oxidation to NO2 over manganese-cerium mixed oxides[J]. Catalysis Today,2015,258:205-213.
[27]? KANTCHEVA M. Identification,stability,and reactivity of NOx species adsorbed on titania-supported manganese catalysts[J]. Journal of Catalysis,2001,204(2):479-494.
[28]? WU Z,JIANG B,LIU Y,et al. DRIFT study of manganese/ titania-based catalysts for low-temperature selective catalytic reduction of NO with NH3[J]. Environmental Science & Technology,2007,41(16):5812-5817.
[29]? TANG N,LIU Y,WANG H Q,et al. Mechanism study of NO catalytic oxidation over MnOx/TiO2 catalysts[J]. The Journal of Physical Chemistry C,2011,115(16):8214-8220.
[30]? MENG D M,ZHAN W C,GUO Y,et al. A highly effective catalyst of Sm-MnOx for the NH3-SCR of NOx at low temperature:promotional role of Sm and its catalytic performance[J]. ACS Catalysis,2015,5(10):5973-5983.
[31]? KURTIKYAN T S,FORD P C. FTIR and optical spectroscopic studies of the reactions of heme models with nitric oxide and other NOx in porous layered solids[J]. Coordination Chemistry Reviews,2008,252(12/13/14):1486-1496.
猜你喜歡
英語世界(2023年10期)2023-11-17 09:18:18
甘肅教育(2020年14期)2020-09-11 07:57:42
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
汽車觀察(2018年10期)2018-11-06 07:05:26
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
少兒科學周刊·少年版(2015年1期)2015-07-07 17:15:12
時代英語·高二(2015年1期)2015-03-16 00:08:11
