一測多評法同時測定扶正固本顆粒中6種成分的含量
2021-02-21 08:18:19仝立國牛艷艷王若瑜吉海杰宋美卿馮瑪莉夏召弟汪欣文
中國藥房 2021年2期
仝立國 牛艷艷 王若瑜 吉海杰 宋美卿 馮瑪莉 夏召弟 汪欣文


摘 要 目的:建立扶正固本顆粒中2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、黃芩苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素等6種成分含量的測定方法。方法:采用高效液相色譜法。色譜柱為Dikma Diamonsil C18,流動相為乙腈-0.1%磷酸水溶液(梯度洗脫),檢測波長為275 nm(0~8 min)、320 nm(8~9 min)和275 nm(9~33 min),流速為1.0 mL/min,柱溫為25 ℃,進樣量為10 μL。以黃芩苷為基準物,采用多點校正法和斜率校正法分別計算另外5種成分的相對校正因子(fk/s),并采用保留時間差值法對待測成分進行色譜峰定位,比較上述兩種一測多評法所得計算值與外標法實測值的差異。結果:2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、黃芩苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素檢測進樣量的線性范圍分別為0.053~2.12、0.163~6.52、0.059~2.36、0.021 6~0.864、0.03~1.2、0.021~0.84 μg(r>0.999),精密度、穩定性(12 h)、重復性試驗的RSD均小于3%;平均加樣回收率為98.72%~99.82%(RSD為0.89%~1.24%,n=9)。以黃芩苷為基準物,2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素多點校正法的fk/s分別為1.172、0.528、1.479、1.820、2.534,斜率校正法的fk/s分別為1.234、0.550、1.559、1.939、2.664;3種方法測得10批扶正固本顆粒中6種成分含量的RSD為0.29%~2.77%(n=10);兩種一測多評法測得結果與外標法的Pearson相關系數均不低于0.999 9(P<0.001)。結論:成功建立了可用于同時測定扶正固本顆粒中6種成分含量的一測多評法。
關鍵詞 一測多評法;扶正固本顆粒;2,3,5,4′-四羥基二苯乙烯基葡萄糖苷;黃芩苷;淫羊藿苷;漢黃芩苷;黃芩素;漢黃芩素;含量
ABSTRACT? ?OBJECTIVE: To establish the method for content determination of 6 components in Fuzheng guben granules, such as 2,3,5,4′-tetrahydroxystilbene glucoside,baicalin,icariin,scutellarin,baicalein and wogonin. METHODS: HPLC method was adopted. The determination was performed on Dikma Diamonsil C18 column with mobile phase consisted of? acetonitrile-0.1% phosphoric acid aqueous solution (gradient elution) at the flow rate of 1.0 mL/min. The detection wavelengths were set at 275 nm (0-8 min), 320 nm(8-9 min) and 275 nm(9-33 min). The column temperature was set at 25 ℃, and sample size was 10 μL. With baicalin as reference material, the relative correction factors (fk/s) of other five components were calculated by multi-point correction method and slope correction method; the retention time difference method was used to locate the chromatographic peaks; the calculation values obtained by above 2 QAMS were compared with measured values of external standard method. RESULTS: The linear range of 2,3,5,4′-tetrahydroxystilbene glucoside, baicalin, icariin, scutellarin, baicalein and wogonin were 0.053-2.12, 0.163-6.52, 0.059-2.36, 0.021 6-0.864, 0.03-1.2, 0.021-0.84 μg(r>0.999), respectively. RSDs of precision, stability (12 h) and reproducibility tests were all lower than 3%. Average recoveries were 98.72%-99.82%(RSDs were 0.89%-1.24%,n=9). Using baicalin as reference material, fk/s of multi-point correction method for 2,3,5,4′-tetrahydroxystilbene glucoside, icariin, scutellarin, baicalein and wogonin were 1.172, 0.528, 1.479, 1.820 and 2.534, respectively; fk/s of slope correction method were 1.234, 0.550, 1.559, 1.939, 2.664. RSDs of 6 components in 10 batches of Fuzheng guben granules by 3 methods were 0.29%-2.77%(n=10), respectively. Pearson correlation coefficient was not lower than 0.999 9 (P<0.001) in measured values between QAMS and external standard method. CONCLUSIONS: QAMS method is established successfully for simultaneous determination of 6 components in Fuzheng guben granules.
KEYWORDS? ?QAMS; Fuzheng guben granules; 2,3,5,4′-tetrahydroxystilbene glucoside; Baicalin; Icariin; Scutellarin; Bai- calein; Wogonin; Content
中圖分類號 R944.2+7;R927.2 文獻標志碼 A 文章編號 1001-0408(2021)02-0225-06
DOI 10.6039/j.issn.1001-0408.2021.02.17
扶正固本顆粒由黃芩、淫羊藿、何首烏等8味中藥組成,具有益氣養陰、涼血解毒的功效,可作為食管癌以及胃寒氣陰兩虛兼熱毒癥患者放、化療的合并用藥,其制劑標準被收載于國家藥品監督管理局國家藥品標準[WS-5250(B-0250)-2002]中。目前,扶正固本顆粒多用于腫瘤患者的輔助治療。雖然,中藥并非是公認的腫瘤治療的有效手段,但隨著中藥補益作用和抗腫瘤作用研究的逐漸深入,其在腫瘤治療中的地位越來越高,應用也日益廣泛[1-2]。扶正固本顆粒現行質量標準中含量測定僅以黃芩苷單一成分為指標[3],難以全面控制其質量,而采用傳統方法對多成分同時進行測定又會使檢測成本大幅增加。一測多評法在控制檢測成本的同時,對多成分進行了控制,可以全面評價中藥及其制劑的質量[4]。本課題組前期采用網絡藥理學方法對扶正固本顆粒藥效物質基礎進行了分析,其有效成分主要集中于黃芩、淫羊藿、何首烏、女貞子以及茜草中。基于此,本研究擬選擇黃酮類、蒽醌類、裂環環烯醚萜苷類以及二苯乙烯苷類等成分作為待測指標,采用一測多評法進行含量測定,旨在為更全面控制扶正固本顆粒的質量提供方法依據。
1 材料
1.1 儀器
本文所用實驗儀器有:Aglient 1200型高效液相色譜儀(包括真空脫氣機、四元泵、自動進樣器、柱溫箱、二極管陣列檢測器、Chemstation B.04.02型色譜工作站,美國Aglient公司)、XS105型分析天平(瑞士Mettler Toledo公司)、明澈-D24UV型超純水器(美國Millipore公司)、S60H型超聲波清洗機(德國Elma公司)。
1.2 藥品與試劑
扶正固本顆粒(批號20160603、20161104、20170806、20170401、20171012、20180302、20180601、20181002、20190301、20190603,規格15 g/袋)和黃芩、淫羊藿、女貞子、何首烏、地黃、茜草、黃精、人參藥材均由山西振東開元制藥有限公司提供,藥材經山西省藥品檢驗所高天愛主任藥師鑒定并確定為真品。
黃芩苷對照品(批號110715-201318,純度93.3%)、黃芩素對照品(批號111595-200604,純度100%)、淫羊藿苷對照品(批號110737-201516,純度94.2%)、漢黃芩素對照品(批號111514-201706,純度100%)、漢黃芩苷對照品(批號112002-201702,純度98.5%)、2,3,5,4′-四羥基二苯乙烯基葡萄糖苷對照品(批號110844-201109,純度94.7%)均購自中國食品藥品檢定研究院;甲醇、乙腈均為色譜純,其余試劑均為分析純,水為純凈水。
2 方法與結果
2.1 色譜條件
以Dikma Diamonsil C18(250 mm×4.6 mm,5 μm)為色譜柱;以乙腈(A)-0.1%磷酸水溶液(B)為流動相進行梯度洗脫(0~8 min,20%A→22.5%A;8~18 min,22.5%A→35%A;18~22 min,35%A;22~26 min,35%A→55%A;26~30 min,55%A;30~31 min,55%A→20%A;31~33 min,20%A);檢測波長為275 nm(0~8 min)、320 nm(8~9 min)和275 nm(9~33 min);流速為1.0 mL/min;柱溫為25 ℃;進樣量為10 μL。
2.2 供試品溶液制備
取扶正固本顆粒約2 g,精密稱定,置于具塞錐形瓶中,精密加入80%甲醇50 mL,稱定質量,超聲(功率250 W,頻率40 kHz)處理30 min,放冷至室溫,再次稱定質量,用80%甲醇補足減失的質量,搖勻,濾過,取續濾液,即得。
2.3 對照品溶液制備
分別稱取2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、黃芩苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素對照品各適量,置于同一量瓶中,用80%甲醇溶解并稀釋,制成質量濃度分別為106、326、118、43.2、60.0、42.0 μg/mL的混合對照品溶液,備用。另稱取2,3,5,4′-四羥基二苯乙烯基葡萄糖苷對照品15.93 mg、黃芩苷對照品34.3 mg、淫羊藿對照品10.81 mg、黃芩素對照品13.61 mg,分別置于10 mL量瓶中;再稱取漢黃芩苷對照品21.04 mg、漢黃芩素對照品18.4 mg,分別置于20 mL量瓶中;均用80%甲醇溶解并稀釋至刻度,制成單一對照品貯備液,備用。
2.4 陰性對照溶液制備
按處方比例和制劑工藝分別制備不含黃芩、淫羊藿、何首烏及以上3個都不含的陰性樣品,再按“2.2”項下方法制備成相應的陰性對照溶液,備用。
2.5 專屬性考察
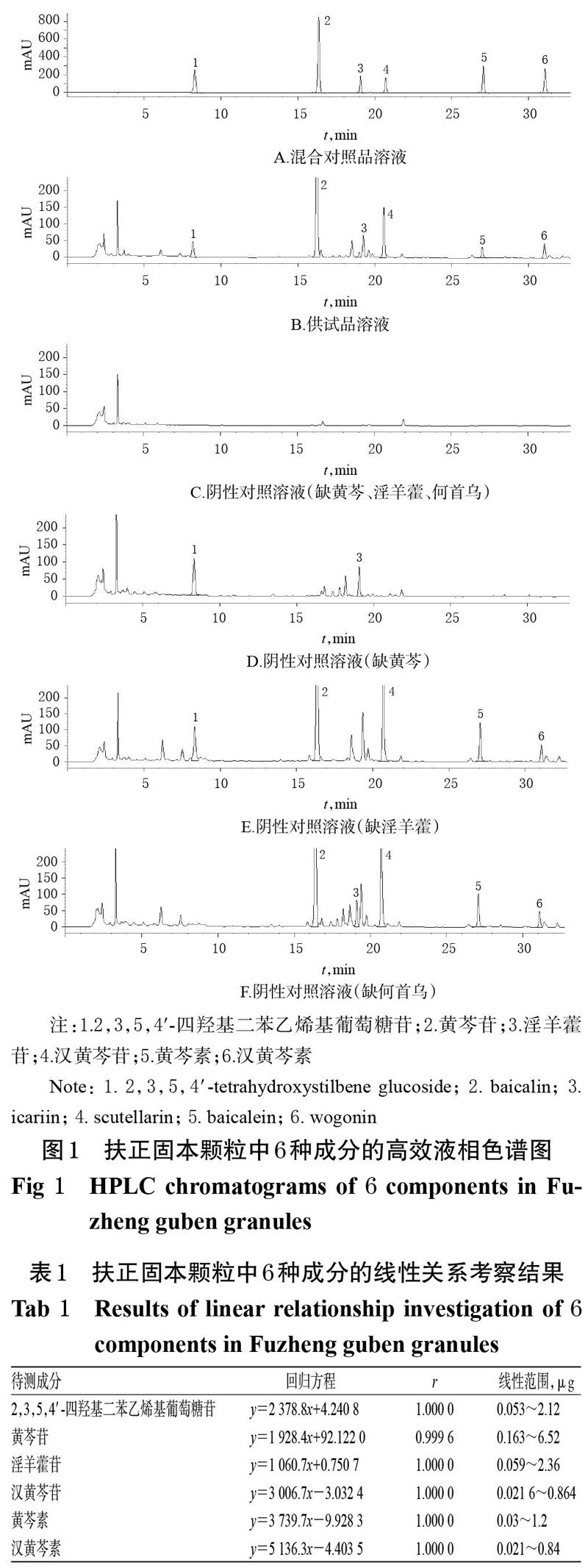
分別精密吸取上述混合對照品溶液、供試品溶液(批號20170806)和各陰性對照溶液適量,按“2.1”項下色譜條件進樣測定,記錄色譜圖。結果,2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、黃芩苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素的保留時間分別為8.298、16.367、19.063、20.710、27.061、31.088 min,分離度均大于1.5,陰性對照溶液在相應位置無干擾,詳見圖1。
2.6 方法學考察
2.6.1 線性關系考察 分別精密吸取混合對照品溶液0.5、1.0、2.0、5.0、10.0、15.0、20.0 μL,按照“2.1”項下色譜條件進樣測定,記錄峰面積。以峰面積(y)為縱坐標、進樣量(x,μg)為橫坐標繪制標準曲線。結果,在各自線性范圍內,各待測成分進樣量與峰面積均呈良好的線性關系(r>0.999),結果見表1。
2.6.2 精密度試驗 精密吸取混合對照品溶液10 μL,按“2.1”項下色譜條件連續進樣6次,連續測定3 d,記錄峰面積并分別計算各待測成分峰面積的RSD。結果,2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、黃芩苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素峰面積的日內RSD分別為0.19%、0.17%、0.19%、0.18%、0.11%、0.09%(n=6),日間RSD分別為0.20%、0.11%、0.18%、0.10%、0.11%、0.14%(n=3),表明儀器精密度良好。
2.6.3 穩定性試驗 取同一批次扶正固本顆粒(批號20170806),按“2.2”項下方法制備供試品溶液,于室溫下放置0、1.5、3.5、5、7、8.5、10.5、12 h時按“2.1”項下色譜條件進樣測定,記錄峰面積并分別計算各待測成分峰面積的RSD。結果,2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、黃芩苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素峰面積的RSD分別為1.66%、0.26%、0.90%、1.69%、0.72%、0.53%(n=8),表明供試品溶液在室溫下放置12 h內穩定性良好。
2.6.4 重復性試驗 取同一批次扶正固本顆粒(批號20170806),共6份,按“2.2”項下方法制備供試品溶液,再按“2.1”項下色譜條件進樣測定,記錄峰面積并按外標法計算含量。結果,2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、黃芩苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素含量的RSD分別為2.25%、1.56%、2.85%、1.89%、2.91%、2.12%(n=6),表明方法重復性良好。
2.6.5 加樣回收率試驗 取同一批次扶正固本顆粒(批號20170806),共9份,3份1組,每份約1 g,分別精密加入“2.3”項下2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、黃芩苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素單一對照品貯備液適量,加入量為供試品中各待測成分已知量的80%、100%、120%;隨后精密加入80%甲醇適量,使總體積為50 mL,按“2.2”項下方法處理后,再按“2.1”項下色譜條件進樣測定,記錄峰面積并計算回收率,結果見表2。
2.6.6 耐用性試驗 分別考察流動相各梯度點比例變化±5%、水相磷酸濃度變化±0.05%、柱溫變化±5 ℃、檢測波長變化±5 nm、流速變化±20%以及不同色譜柱[Dimak Diamonsil C18、TIANHE Kromasil C18、ELITE SinaChrom C18(均為250 mm×4.6 mm,5 ?m)],其余色譜條件同“2.1”項時分離度和理論板數的變化。結果,各梯度點比例變化±5%、檢測波長變化±5 nm以及采用3根色譜柱時,2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、黃芩苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素色譜峰的分離度均大于1.5,理論板數(分別以各待測成分計)均大于15 000;而在水相磷酸濃度變化為-0.05%、柱溫變化±5 ℃、流速變化±20%時,淫羊藿色譜峰的分離度不能滿足分析要求,其余5個待測成分色譜峰的分離度均大于1.5,理論板數(分別以各待測成分計)均大于15 000。這表明水相磷酸濃度、柱溫以及流速對淫羊藿苷的檢測具有較大影響,即該成分檢測條件要求較為嚴格、耐用性稍差;而其余5個待測成分在上述條件變動的情況下仍具有較好的分離度及理論板數,耐用性較好。
2.7 相對校正因子計算
2.7.1 多點校正法 參考文獻方法[5-6],以多個質量濃度點計算所得校正因子的平均值作為相對校正因子。計算公式為:
兩式中,fk/s為相對校正因子;cs為參照物質質量濃度;As為參照物質峰面積;ck為其他對照組分質量濃度;Ak為其他對照組分峰面積;ck′為待測組分質量濃度;Ak′為待測組分峰面積。應用此法需先獲得一個參照物質的質量濃度cs和峰面積As。
以黃芩苷為基準峰(即參照物質,黃芩苷含量高且與其他色譜峰的分離度良好,下同),取6種待測成分的混合對照品溶液按“2.1”項下色譜條件進樣測定,進樣量分別為0.5、1、2、5、10、15 μL,記錄峰面積,按公式①計算2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素的相對校正因子,結果見表3。
2.7.2 斜率校正法 參考文獻方法[3-4],標準曲線回歸方程y=ax+b中,x=(y-b)/a=y/a-b/a,由于b值通常為誤差所致,在a/b值大于100時,b/a值可以忽略不計,此時可以直接用x=y/a計算含量。校正因子可以二者的斜率a之比直接計算,相對校正因子計算公式為:
兩式中,as為參照物質斜率;ak為其他對照組分斜率。
以黃芩苷為基準峰,將“2.6.1”項下6種待測成分回歸方程的相應數據,代入公式③計算2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素的相對校正因子,結果見表4。
2.8 待測組分色譜峰定位
2.8.1 相對保留時間法 參考文獻方法[7-8],利用相對保留時間及二極管陣列檢測器測定的紫外吸收光譜對各組分進行定位及確認,以各待測組分色譜峰與黃芩苷色譜峰的保留時間之比計算相對保留時間。以黃芩苷為基準峰,計算得“2.6.6”項不同測試條件下2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素的相對保留時間分別為0.526、1.164、1.261、1.643、1.893,其RSD分別為6.52%、0.97%、1.61%、2.66%、2.56%(n=13)。
2.8.2 保留時間差值法 參考文獻方法[7],利用相對保留時間及二極管陣列檢測器測定的紫外吸收光譜對各組分進行定位及確認,以各待測組分色譜峰與黃芩苷色譜峰的保留時間的差值計算相對保留時間。以黃芩苷為基準峰,計算得“2.6.6”項不同測試條件下2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素的相對保留時間分別為-7.886、2.712、4.343、10.697、14.864,其RSD分別為3.21%、3.15%、2.68%、1.35%、1.46%(n=13)。
對比兩種方法結果的RSD值發現,保留時間差值法計算的相對保留時間的精密度總體更高,故后續以保留時間差值法對待測組分進行定位。
2.9 含量測定比較
取10批扶正固本顆粒,按“2.2”項下方法制備供試品溶液,再按“2.1”項下色譜條件進樣測定,分別用外標法和兩種一測多評法測定或計算2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、黃芩苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素的含量,再計算RSD值;以及兩種一測多評法的測定結果分別與外標法的Pearson相關系數(r),上述統計學處理均由SPSS 22.0軟件完成,結果見表5。
由表5結果顯示,兩種一測多評法與外標法所得各待測成分含量的RSD分別為2.77%、2.58%、0.14%、0.51%、0.96%、0.29%,兩者相關性好(r均不低于0.999 9,P<0.001),說明兩種一測多評法與外標法結果基本一致。
3 討論
本研究選擇2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、黃芩苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素等6種成分進行一測多評分析研究,其成分主要來源于黃芩、淫羊藿、何首烏等3味中藥,是扶正固本顆粒的主要藥效成分[9-11]。在前期試驗過程中,本課題組曾對何首烏中的毛蕊花糖苷、大黃素、大黃酸、大黃素甲醚、大黃酚、淫羊藿中的寶藿苷Ⅰ,女貞子中的女貞苷和特女貞苷,茜草中的大葉茜草素、羥基茜草素等成分進行檢測,發現毛蕊花糖苷、女貞苷、特女貞苷、大葉茜草素、羥基茜草素等成分在該色譜條件下未被檢出,大黃素、大黃酸、大黃素甲醚、大黃酚、寶藿苷Ⅰ等成分在該色譜條件下響應較低,故選擇了成分含量相對較高且分離度較好的6種成分。
一測多評法的兩個關鍵問題:(1)對各成分的定位。本文比較了相對保留時間和保留時間差值兩種較為簡單的方法對各成分進行定位的效果。結果顯示,保留時間差值法的RSD值小于相對保留時間法的RSD值,提示保留時間差值法能夠相對更好地對待測成分進行定位;(2)相對校正因子的計算。本文比較了多點校正法、斜率校正法以及外標法對各成分測定的結果。結果,3種方法所得的各成分含量之間無顯著差異,提示多點校正法和斜率校正法計算的相對校正因子均可滿足其余5種成分的定量要求。
隨著中藥現代研究的不斷深入,中藥藥效成分的多樣性和復雜性使得其多指標定量分析成為必然趨勢,然而按照傳統定量模式,勢必存在對照品消耗量大,檢驗成本升高,計算繁瑣、易出現誤差等不利因素[8]。中藥一測多評質量評價模式則克服了以上缺點,尤其適合于企業或一些實驗室對某類長期生產和需重復測定樣品的例行檢測,此法可提升分析檢測的準確性和效率,并可大大地降低分析檢測的成本[7]。本研究借助高效液相色譜法對扶正固本顆粒中6種成分的一測多評方法進行研究,結果表明,通過兩種一測多評法計算出的含量與外標法實際測量的含量相關性好,提示以黃芩苷為參照物質,采用一測多評法測定扶正固本顆粒中2,3,5,4′-四羥基二苯乙烯基葡萄糖苷、淫羊藿苷、漢黃芩苷、黃芩素、漢黃芩素的含量具有良好的準確性和可操作性。該方法可用于相關企業及實驗室的日常檢驗工作。
參考文獻
[ 1 ] 李露露,張嬋,焦士潔,等.扶正固本顆粒治療晚期結直腸癌合并貧血的臨床研究[J].癌癥進展,2017,15(9):1048-1051.
[ 2 ] 陳東基,李小軍,付國翠,等.同期放化療聯合扶正固本顆粒治療中晚期食管癌臨床觀察[J].中成藥,2012,34(3):406-409.
[ 3 ] 朱艷容,李媛媛,倪艷,等.扶正固本顆粒的質量標準研究[J].中國藥房,2011,22(23):2179-2181.
[ 4 ] 王智民,高慧敏,付雪濤,等.“一測多評”法中藥質量評價模式方法學研究[J].中國中藥雜志,2006,31(23):1925- 1928.
[ 5 ] 何兵,楊世艷,張燕.一測多評中待測成分校正和定位的新方法研究[J].藥學學報,2012,47(12):1653-1689.
[ 6 ] 呂曉霞,陳宗良,陳桂茜,等.一測多評法在仙靈骨葆膠囊中多成分檢測的應用研究[J].中草藥,2016,47(24):4374-4378.
[ 7 ] 陳俊,許浚,張靜雅,等.基于一測多評法對延胡索中生物堿類成分的質量控制研究[J].中草藥,2016,47(3):493- 498.
[ 8 ] 羅祖良,仇峰,韋日偉,等.相對校正因子在中藥多指標測定中的應用研究進展[J].中草藥,2012,43(7):1448- 1452.
[ 9 ] 王浩,楊健,周良云,等.何首烏化學成分與藥理作用研究進展[J].中國實驗方劑學雜志,2019,25(13):192-205.
[10] 朱亞南,楊七妹,張碩,等.黃芩苷與黃芩素藥理作用及機制研究進展[J].時珍國醫國藥,2020,31(4):921-925.
[11] 何麗君,江金井,陳豪,等.淫羊藿藥理作用和臨床應用的研究進展[J].中醫臨床研究,2020,12(2):17-20.
(收稿日期:2020-07-27 修回日期:2020-11-17)
(編輯:鄒麗娟)
