
利用CRISPR/Cas9慢病毒載體構建敲除大鼠肝星狀細胞COX-2基因的細胞模型
2021-03-03 02:32:34陽學風易世杰周克兵龍建武
臨床肝膽病雜志 2021年2期
關鍵詞:檢測
彭 敏, 曹 婷, 陽學風, 易世杰, 傅 念, 周克兵, 龍建武
南華大學附屬南華醫(yī)院 消化內科, 湖南 衡陽 421002
肝纖維化是一系列慢性肝損傷導致的病理生理過程[1],它是發(fā)展為肝硬化的必經(jīng)過程。目前認為,肝星狀細胞(HSC)的活化是肝纖維化發(fā)生的中心環(huán)節(jié),因此,通過抑制HSC活化可以達到逆轉肝纖維化的目的[2]。環(huán)氧合酶(cyclooxygenase,COX)是花生四烯酸合成前列腺素的關鍵限速酶[3],包括COX-1、COX-2和COX-3三個亞型,COX-2屬于誘導型酶。國內外大量研究[4]證明,COX-2參與肝損傷炎癥反應,促進肝纖維化發(fā)展病程,也調節(jié)HSC活化及增殖,參與進一步肝損傷。已有相關研究[5-9]報道表明,抑制COX-2表達,能夠下調HSC活化和增殖,可能成為逆轉肝纖維化的策略。目前COX-2和肝纖維化關系的研究主要集中在COX-2抑制劑或siRNA干擾技術上,至今沒有文獻報道完全敲除HSC中COX-2基因對HSC功能的影響及機制。近年來隨著CRISPR/Cas9技術的興起,使通過CRISPR/Cas9技術對COX-2基因進行敲除成為一種新的研究方向。基于CRISPR/Cas9系統(tǒng)相比于RNAi技術更容易獲得穩(wěn)定的表型變化,而且具有設計簡便、易于操作及特異性高的優(yōu)勢[10]。因此本實驗通過構建慢病毒表達載體,利用CRISPR/Cas9技術敲除HSC的COX-2基因,以期獲得能穩(wěn)定傳代的COX-2基因缺陷的細胞模型,為今后研究COX-2基因缺陷的HSC功能及機制提供實驗基礎,為臨床上治療肝纖維化提供新策略。
1 材料與方法
1.1 實驗材料 HSC-T6細胞購自中科院上海細胞庫,293T細胞購自南京科佰生物科技有限公司,Lenti-CAS9-puro質粒、Lenti-NC-EGFP質粒、GV371質粒、p Helper 1.0載體,p Helper 2.0載體、Primer(R&F)、dsDNA oligo、TOP10感受態(tài)均購自上海吉凱基因化學技術有限公司,Lipofectamine TM2000購自美國Invitrogen公司,基因組 DNA 抽提試劑盒購自TIANGEN公司,基因敲除和突變檢測試劑盒購自吉盛醫(yī)學公司。
1.2 實驗方法
1.2.1 COX-2-sgRNA表達載體構建 (1)293T細胞培養(yǎng):①細胞復蘇;②細胞傳代;③細胞凍存。(2)sgRNA靶點序列的選擇與合成:①靶序列選擇。根據(jù)上海吉凱基因化學技術有限公司設計3條COX-2-sgRNA干擾靶點序列,sgRNA-1:5′-AAATGTGACTGTACCCGGAC-3′;sgRNA-2:5′-CATGATTGAATTCCGAAGGA-3′;sgRNA-3:5′-TTTCACCGTAGAATCCAGTC-3′。②寡核苷酸單鏈合成:以上述靶序列為基礎,設計合成3對寡核苷酸單鏈,其兩端加入BbsI位點。(3)引物退火形成帶黏性末端的雙鏈:將引物干粉溶解于退火緩沖液中,95 ℃水浴15 min,冷卻至室溫,稀釋200倍后使用。(4)sgRNA表達載體酶切重組:通過酶切連接反應體系進行酶切連接反應,結束后將形成的重組質粒直接轉化。(5)重組質粒轉化、測序:取n支TOP10感受態(tài),進行培養(yǎng),待菌落長出后,挑取8個大的單克隆菌落于15 ml的液體培養(yǎng)基,送公司進行測序。(6)質粒提取:參照E.Z.N.A Endo-free plasmid mini kit試劑盒說明書標準方法制備,所得DNA溶液-20 ℃保存或直接用于下一步實驗。(7)病毒包裝:①病毒轉染;②病毒的收獲及濃縮;③病毒滴度的測定(熒光法)。使用熒光顯微鏡觀察轉染效果,圖片均使用Pixera Camera圖像分析系統(tǒng)采集。
1.2.2 CRISPR/Cas9慢病毒轉染HSC-T6細胞及活性檢測 (1)準備目的細胞:HSC-T6細胞的復蘇、傳代和凍存同前所述293T細胞的培養(yǎng)方法。(2)細胞轉染:①Lenti-Cas9-puro病毒轉染HSC-T6細胞,構建穩(wěn)定表達Cas9蛋白的HSC-T6細胞,收集細胞標記為HSC-T6-Cas9細胞,進行下一步實驗。②Real-time PCR檢測Cas9病毒轉染HSC-T6細胞后的表達情況:提取總RNA,逆轉錄獲得cDNA(按照Promega公司提供的逆轉錄試劑盒操作步驟進行)將得到的產(chǎn)物cDNA -20 ℃保存?zhèn)溆茫詈筮M行Real-time PCR,LV-Cas9-Puro基因的引物由廣州市銳博生物科技有限公司設計合成,引物序列如表1,按比例配置反應體系(12 μl體系),反應結束后通過制作熔解曲線確認為單峰曲線,基因表達量用相對定量分析法。(3) HSC-T6-Cas9細胞的凍存、復檢:qPCR檢測結果合格后進行HSC-T6-Cas9細胞凍存,至少凍存6支,凍存2~3 d,任意復蘇1支,顯微鏡下觀察復蘇細胞狀態(tài)。(4)Lenti-COX-2-sgRNA-EGFP病毒轉染HSC-T6-Cas9細胞:①在上一步實驗的細胞基礎上,將細胞分為3組,分別為KO1組(Lenti-COX-2-sgRNA-1病毒轉染組)、KO2組(Lenti-COX-2-sgRNA-2病毒轉染組)、KO3組(Lenti-COX-2-sgRNA-3病毒轉染組),加入相對應的Lenti-COX-2-sgRNA-1、Lenti-COX-2-sgRNA-2、Lenti-COX-2-sgRNA-3病毒進行感染。同時設立NC組(Lenti-NC病毒轉染組)、CON組(HSC-T6細胞)。②感染72 h左右,在熒光顯微鏡下觀察細胞中綠色熒光的表達狀況,估計熒光率;③分別用含1 μg/ml puromycin的培養(yǎng)液培養(yǎng)傳代細胞,得到COX-2基因敲除的HSC-T6-COX-2-/-混合細胞,分別標記為HSC-T6-COX-2-/--1細胞、HSC-T6-COX-2-/--2細胞、HSC-T6-COX-2-/--3細胞,并收集細胞做下一步檢測。(5)Lenti-NC-EGFP病毒轉染HSC-T6細胞:根據(jù)MOI值,感染Lenti-NC-EGFP病毒,在熒光顯微鏡下觀察細胞中綠色熒光的表達情況,估計熒光率,收集細胞做下一步檢測,標記為HSC-T6-NC細胞。
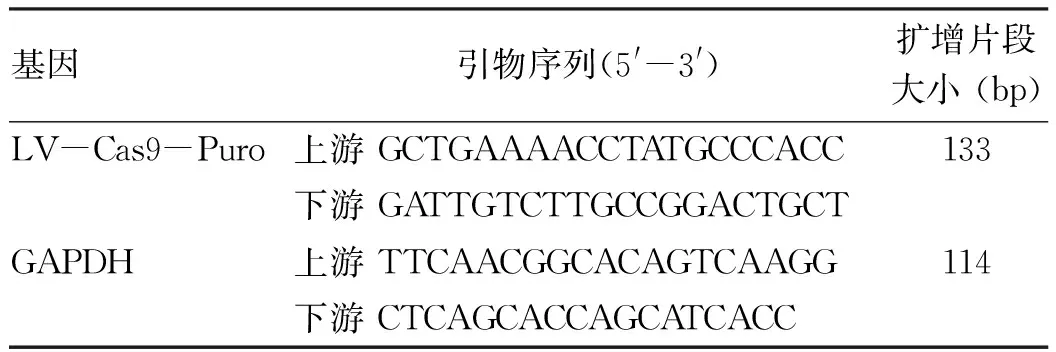
表1 LV-Cas9-Puro基因的引物序列
1.2.3 CRISPR/Cas9系統(tǒng)對COX-2靶點的敲除活性檢測 (1)混合細胞克隆pool檢測:①提取各基因組DNA;②PCR引物設計,按照引物設計通用原則,由上海吉凱公司設計合成(表2);③按比例配置反應體系,PCR反應程序反應后獲得雜交DNA產(chǎn)物;④PCR結束后,取3 μl進行電泳檢測。(2)Cruiser酶切檢測:在滅菌PCR管中配制PCR產(chǎn)物2~3 μl、Detecase Buffer 2 μl、Detecase 1 μl、ddH2O至10 μl,45 ℃反應20 min后,立即向上述10 μl體系內加入2 μl終止液(Stop Buffer),然后進行2%瓊脂糖凝膠電泳檢測。(3)蛋白水平檢測:①蛋白抽提;②SDS-PAGE;③免疫印跡(濕轉);④抗體雜交;⑤X光顯影。
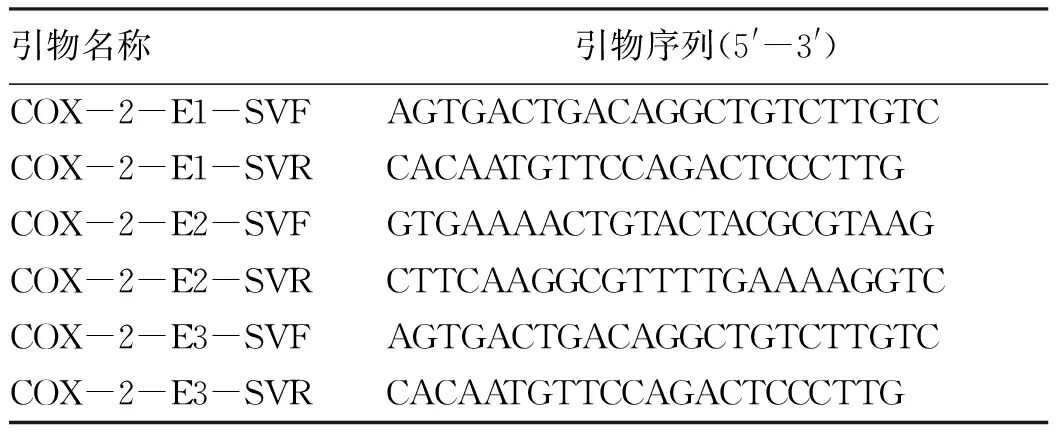
表2 PCR引物序列
2 結果
2.1 COX-2-sgRNA表達載體構建 根據(jù)公司設計3條sgRNA,在其兩端加入BbsI位點,化學合成設計序列并退火形成帶有黏性末端的雙鏈,將其連接到經(jīng)BbsI限制性核酸內切酶切割形成的線性化GV371表達載體上,經(jīng)轉化擴增載體的表達,并將轉化子測序驗證。測序結果顯示,COX-2-sgRNA-1、2、3序列均成功插入目的載體,且序列完全正確。證實COX-2-sgRNA表達載體構建成功(圖1)。

注:a,COX-2-sgRNA-1; b, COX-2-sgRNA-2; c,COX-2-sgRNA-3。
2.2 病毒滴度的測定 在倒置顯微鏡及熒光顯微鏡下觀察加入病毒不同稀釋倍數(shù)的293T細胞熒光表達情況顯示,熒光細胞數(shù)量隨病毒稀釋倍數(shù)增加而減少,按照滴度計算公式:細胞數(shù)量×感染效率/感染體積,計算出COX-2-sgRNA-1的病毒滴度為8×108,COX-2-sgRNA-2、COX-2-sgRNA-3的病毒滴度為1×109(圖2)。

圖2 不同稀釋倍數(shù)COX-2-sgRNA慢病毒感染293T細胞的熒光表達情況(×100)
2.3 細胞轉染實驗
2.3.1 puromycin篩選HSC-T6-Cas9細胞 Lenti-Cas9-puro病毒為抗性基因標記,如果轉染入細胞,于感染48~72 h后,換用含puromycin的培養(yǎng)基篩選細胞,細胞存活即為陽性感染,圖3為puromycin篩選前后HSC-T6-Cas9細胞狀態(tài)及熒光表達情況。與CON組對比,篩選前后HSC-T6-Cas9細胞狀態(tài)好,未出現(xiàn)大量死亡現(xiàn)象。

注:左右分別是同一視野下光學顯微鏡圖片和熒光顯微鏡圖片。a,CON組;b,puromycin篩選前HSC-T6-Cas9細胞;c,puromycin篩選后HSC-T6-Cas9細胞。
2.3.2 Real-time PCR檢測Cas9病毒轉染HSC-T6細胞后的表達情況 收集轉染獲得的HSC-T6-Cas9細胞以及CON組細胞,分別提取LV-Cas9-Puro mRNA,利用Real-time PCR檢測獲得各組基因循環(huán)閾值,并繪制各基因在各組樣品中的相對定量直方圖。結果顯示:CON組和HSC-T6-Cas9細胞組目的基因的相對表達量分別為1.00±0.02、541.93±105.76,提示HSC-T6-Cas9細胞中LV-Cas9-Puro mRNA表達高于CON組細胞,差異具有統(tǒng)計學意義(t=12.995,P<0.01)(圖4、5)。

圖4 擴增曲線
2.3.3 HSC-T6-Cas9細胞凍存后復檢 經(jīng)Real-time PCR檢測HSC-T6-Cas9細胞中LV-Cas9-Puro mRNA表達豐度明顯高于CON組,將HSC-T6-Cas9細胞凍存2~3 d后復蘇,在顯微鏡下觀察,結果顯示:復蘇細胞狀態(tài)較好(圖6),提示凍存細胞合格,穩(wěn)定表達Cas9蛋白的HSC-T6細胞構建成功。

圖5 熔解曲線

注:左右分別是同一視野下光學顯微鏡圖片和熒光顯微鏡圖片。
2.3.4 Lenti-COX-2-sgRNA-EGFP病毒轉染HSC-T6-Cas9細胞 COX-2-sgRNA質粒為熒光標記,如果轉染入細胞,于感染72 h 左右,熒光顯微鏡下觀察,細胞會出現(xiàn)綠色熒光。隨機選擇10個視野,計算陽性細胞與總細胞之比作為質粒轉染效率指標。圖7為各組病毒轉染72 h熒光顯微鏡圖片。KO1、KO2、KO3、NC組細胞均可見綠色熒光,轉染效率在80%以上,說明病毒轉染成功,并且puromycin對細胞無殺害作用。CON組未見綠色熒光出現(xiàn)。
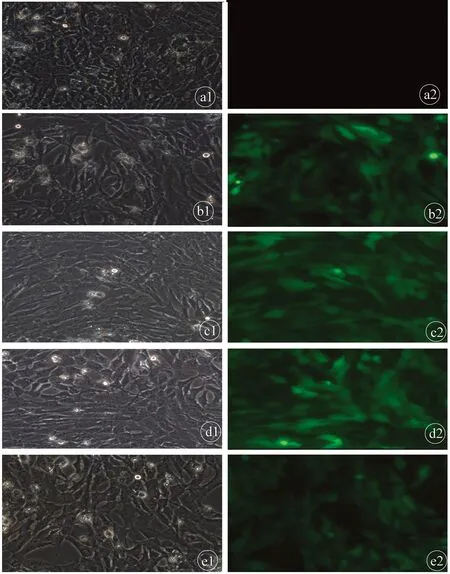
注:左右分別是同一視野下光學顯微鏡圖和熒光顯微鏡圖。a,CON組;b,KO1組;c,KO2組;d,KO3組;e,NC組。
2.4 CRISPR/Cas9系統(tǒng)對COX-2靶點的敲除活性檢測
2.4.1 Cruiser酶切檢測 為了研究CRISPR/Cas9系統(tǒng)對靶點的敲除作用,本實驗通過將構建好的Cas9蛋白表達載體和sgRNA表達載體轉染細胞。72 h后提取基因組,在敲除靶點上下游設計引物,通過PCR擴增出包含敲除靶點的一條目的片段,取部分行電泳檢測,利用Cruiser酶切檢測其敲除效率(圖8)。從結果可以看出,針對COX-2靶基因所設計的CRISPR/Cas9慢病毒表達載體能在靶點起作用。作用于KO1組靶點序列引物COX-2-E1-SVF/R,擴增片段長為369 bp,酶切片段長分別為120 bp、249 bp;KO2組靶點序列引物COX-2-E2-SVF/R,擴增片段長為383 bp,酶切片段長分別為251 bp、132 bp;KO3組靶點序列引物COX-2-E3-SVF/R,擴增片段長為369 bp,酶切片段長分別為123 bp、246 bp。而CON組與NC組未出現(xiàn)酶切條帶,提示無突變位點。KO1、KO2、KO3靶點有活性,且KO2活性較佳,選用KO2組進行后續(xù)蛋白水平驗證。
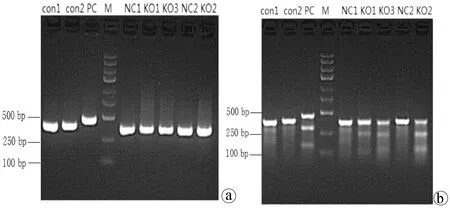
注:a,酶切前;b,Cruiser酶切后。
2.4.2 蛋白水平驗證 分別于轉染72 h、96 h提取CON、NC、KO組中的COX-2蛋白,利用Western-Blot檢測COX-2蛋白質表達情況。結果顯示:KO組與CON組、NC組比較,COX-2蛋白質表達明顯下調,灰度值差異具有統(tǒng)計學意義(P值均<0.01)(表3,圖9、10)。

表3 轉染72 h、96 h各組COX-2蛋白質相對表達情況
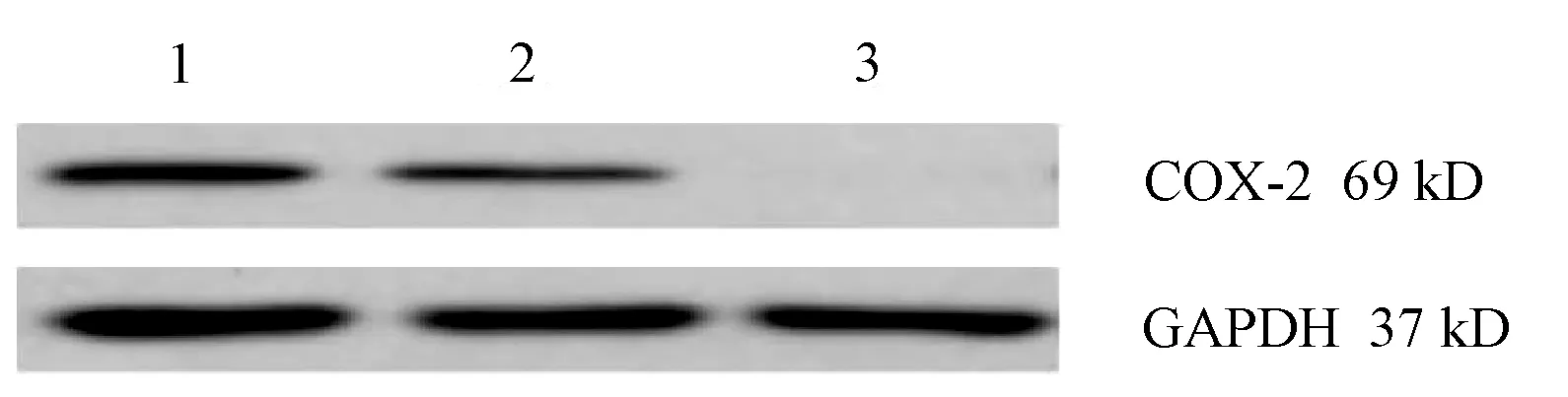
注:1,CON組;2,NC組;3,KO組。

注:1,CON組; 2,NC組; 3,KO組。
3 討論
HSC活化被普遍認為是肝纖維化發(fā)生的關鍵環(huán)節(jié),是肝纖維化治療的重要靶點,將抗纖維化藥物靶向遞送至HSC,減少藥物非特異性作用引起的副作用,是目前抗纖維化藥物開發(fā)的重要策略[11]。同時COX-2參與調節(jié)HSC活化和增殖,參與肝纖維化發(fā)病進程。最新研究[12]表明,COX-2的結構和構象的變化可動態(tài)監(jiān)測肝病的進展,但目前COX-2和肝纖維化關系的研究主要集中在COX-2抑制劑或RNAi技術上,這些技術存在特異性不高、不能獲得穩(wěn)定傳代的細胞株等缺陷,因此需要一種高效、特異性強、穩(wěn)定的基因編輯技術,人為敲除細胞內相關基因的部分片段,從而獲得能穩(wěn)定傳代的COX-2基因缺陷的HSC進行后期的功能研究。
慢病毒以RNA為模板,先在反轉錄酶的作用下合成cDNA,再以此為模板合成雙鏈DNA,通過整合酶作用,整合在宿主細胞的染色體上,可以長期表達[13]。CRISPR/Cas9系統(tǒng)作為第3代基因組編輯工具,與第1代鋅指核糖核酸酶和第2代轉錄激活樣效應因子核酸酶相比,該系統(tǒng)由sgRNA識別靶位點,具有低成本、簡便、精確、高效等優(yōu)勢。CRISPR/Cas9技術為各物種和多種細胞的基因組編輯開拓了選擇途徑。到目前為止,CRISPR/Cas9技術已經(jīng)成功用于小鼠、斑馬魚、酵母、擬南芥、小麥、大米以及哺乳動物細胞等。
最初人們是利用電穿孔、核轉染與脂質體的方式將Cas9和sgRNA轉入細胞內[14-16],但利用電穿孔的方法進行轉染對細胞損傷較大,達不到理想的轉染效果。病毒載體如逆轉錄病毒載體、慢病毒載體[17]、腺病毒載體等能提高基因組編輯的效率,在體外應用比較廣泛[18]。所以本實驗采用慢病毒載體將sgRNA、Cas9導入HSC-T6細胞,構建COX-2基因敲除的HSC-T6細胞模型。本實驗成功構建了3個慢病毒載體經(jīng)測序驗證,結果顯示,COX-2-sgRNA-1、2、3序列均成功插入目的載體,且序列完全正確。證實COX-2-sgRNA表達載體構建成功。使用熒光定量法檢測出各組病毒滴度均在1×108以上。將Cas9慢病毒分別轉染至HSC-T6細胞,使用puromycin篩選、Real-time PCR檢測以及凍存后復檢,結果顯示,利用CRISPR/Cas9技術,即使不進行單克隆篩選,也能永久敲除COX-2基因,相比較RNAi而言更有優(yōu)勢。
本實驗構建了針對COX-2靶基因的CRISPR/Cas9慢病毒表達載體,成功轉染HSC-T6細胞,獲得能穩(wěn)定傳代的Cas9蛋白的HSC-T6細胞和COX-2基因缺陷的HSC-T6-COX-2-/-細胞,為后期的功能研究提供良好的工具和手段,為臨床上治療肝纖維化提供新策略。在此基礎上,本課題組會繼續(xù)研究COX-2基因敲除后HSC-T6細胞的增殖、凋亡、衰老、自噬等功能的變化,并探討其作用機制,爭取將COX-2對HSC-T6細胞作用的具體機制闡述清楚。
利益沖突聲明:本研究不存在研究者、倫理委員會成員、受試者監(jiān)護人以及與公開研究成果有關的利益沖突,特此聲明。
作者貢獻聲明:彭敏、曹婷、陽學風負責課題設計,資料分析,撰寫論文;易世杰、周克兵、龍建武參與收集數(shù)據(jù),修改論文;傅念、陽學風負責擬定寫作思路,指導撰寫文章并最后定稿。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2019年9期)2019-11-25 07:34:36
中學生數(shù)理化·七年級數(shù)學人教版(2019年9期)2019-11-25 07:34:34
中學生數(shù)理化·七年級數(shù)學人教版(2019年12期)2019-05-21 02:53:50
中學生數(shù)理化·七年級數(shù)學人教版(2019年12期)2019-05-21 02:53:48
