聚合物-金屬有機框架材料的研究進展
2021-03-06 03:00:28張家赫原野王明王志王紀孝
化工學報 2021年2期
關鍵詞:結構
張家赫,原野,王明,王志,王紀孝
(1 天津大學化工學院化學工程研究所,天津市膜科學與海水淡化技術重點實驗室,化學工程聯合國家重點實驗室,天津化學化工協同創新中心,天津300350; 2 濱州學院化工與安全學院,山東省工業污水資源化工程技術研究中心,山東濱州256600)
引 言
金屬有機框架(metal?organic frameworks,MOFs)又稱為多孔配位聚合物(porous coordination polymers,PCPs),在近二十年里受到了廣泛的關注。MOFs 是由金屬離子和有機配體組成的多孔雜化材料[1?2],具有比表面積大、多孔結構規則且多樣和孔徑可調等多種優勢[3?5]。然而,與有機聚合物不同,MOF 晶體的可塑性與力學性能均較差[6]。例如,MOF 催化劑在催化反應中極易破碎并堵塞管道;MOF 膜力學性能不高,極易破碎,使用過程中膜易產生缺陷[7?9]。
為解決上述問題,研究者們起初通過表面接枝改性[10?13]和孔道內封裝聚合物鏈[14?18]等方式,利用柔性的聚合物材料對剛性的MOF 材料進行簡單修飾。柔性的有機聚合物具有良好的可加工性,將其引入MOF 材料中可以在一定程度上結合二者的優勢,改善MOFs 的力學性能,提高MOFs 的穩定性與可加工性。例如,在催化或藥物輸送等領域,在MOFs 表面接枝酯類[19?23]、聚N?異丙基丙烯酰胺(PNIPAM)[12,24?25]和聚乙二醇(PEG)[26?28]等聚合物制備MOFs/聚合物復合材料,能夠提高MOF 顆粒在溶液中的分散性、穩定性和力學性能,有效改善由分散性和力學性能差引起的MOF 顆粒破碎或分散不均勻等問題。
然而,由于聚合物與MOFs 之間的相容性較差,僅僅將聚合物作為“客體”引入到MOF 材料中很難實現真正的“無縫融合”[29]。當聚合物鏈段與MOF晶體的金屬簇直接接觸時,由于兩者結構差異大,導致聚合物鏈段和金屬簇親和性不佳;當聚合物鏈段和MOF 晶體的有機配體直接接觸時,有機配體的剛性特征使得配體中的絕大多數原子很不活潑,有機配體與聚合物鏈段的結合力也較弱。因此,MOFs/聚合物復合材料在使用中往往會出現聚合物脫落的現象,導致材料性能下降,使用壽命降低。此外,在MOF 材料表面簡單地混合或接枝聚合物極易導致孔道堵塞,比表面積降低,吸附或反應位點減少等[30]。
為充分結合MOFs 出色的功能性與聚合物的可加工性,將聚合物作為多孔材料的一部分,通過直接或間接的方法合成一種三維且高度多孔的材料——聚合物?金屬有機框架(polymer?metal?organic frameworks, polyMOFs),無疑是一種最佳的途徑。polyMOF 材料中聚合物與MOF 材料是一個整體,其兼具MOFs 與聚合物的優勢,即具有大比表面積、規則可調的孔徑結構以及聚合物的柔性與可加工性、良好的穩定性等特性。
近年來,polyMOFs 作為一種易于加工的新型功能性材料備受關注,諸多優勢使其在生物醫學[31?33]、催化[34?36]、氣體分離[37?40]等領域具有廣闊的應用前景。在Web of Science 中檢索并統計了有關polyMOF 材料的研究論文,其結果如圖1 所示。可以發現,自2008 年以來,polyMOFs 領域發表論文數逐年增加,其研究迅速發展。本文介紹了polyMOF材料的研究現狀,綜述了polyMOF 材料的合成方法和存在的問題,并展望了其未來的發展趨勢。
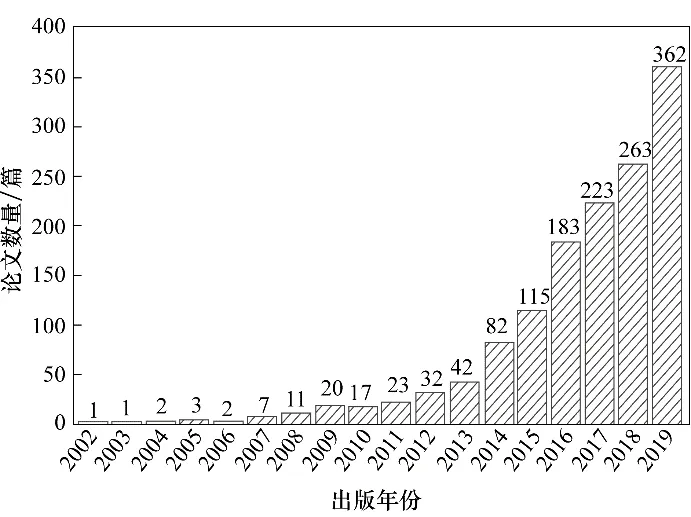
圖1 2000—2019年發表的與polyMOFs相關的論文數(數據來源:Web of Science;搜索的關鍵詞為:MOFs,polymers,polyMOFs)Fig.1 The number of papers on polyMOFs published from 2000 to 2019(Database source:Web of Science;search using keywords:MOFs,polymers,polyMOFs)
1 合成后聚合
合成后聚合(postsynthetic polymerization,PSP)是指在MOF 框架已經形成的基礎上,通過系列后修飾方法將聚合物鏈段或者聚合物網狀結構嵌入MOFs[41]。PSP法通常在無溶劑或者良性溶劑條件下進行,反應過程幾乎不產生副產物,可以高效便捷地合成polyMOF 材料。Zhang 等[42]提出合成后聚合的概念,他們通過光聚合使改性后的UiO?66?NH2納米粒子與丙烯酸酯單體共聚,將MOF 晶體與聚合物鏈有序地組裝在一起生成了交聯的聚合物薄膜,此PSP 方法提供了一種簡單高效的原位策略,可用于制備基于MOFs 的共價連接的混合基質膜[43?45]。目前用于合成polyMOFs 的PSP 技術包括點擊化學法、MOFs孔道內的聚合和表面接枝法等[46?48]。
1.1 點擊化學
點擊化學(click chemistry)反應是Decker 等[49]于2001 年提出的概念,它是指利用小單元的化學原料,通過快速高效、可靠的化學反應來實現碳雜原子之間的連接,最典型的點擊化學反應是銅催化的疊氮?炔基Husigen 環加成反應(copper?catalyzed azidealkyne cycloaddition,CuAAC)[50]。點擊化學因其反應條件溫和、反應速率高、產物易分離等特點而成為制備polyMOF材料的主要方法之一。
Goto 等[51]利用具有疊氮化物官能團的有機配體制備了MOF?16,并證實了可以通過炔烴的CuAAC對合成的MOF進行修飾。在此基礎上,Ishiwata等[52]進一步研究發現含有兩個及以上疊氮基團的有機配體,與多個炔烴位點反應時,能夠在MOF 晶格中發生多次點擊反應,將配體彼此連接,直至聚合產生交聯的MOF(CLM),實現了MOF 內部有機配體的交聯(圖2)。Tsotsalas 等[53]采用類似的方法,使用2,2?二疊氮基?4,4?苯乙烯二羧酸(DA?SBDC)制備了高度有序的SURMOF,然后利用無銅(I)催化的點擊化學反應,將SURMOF 中的疊氮基與炔烴交聯得到聚合物網絡。上述研究證明了MOFs 中的配體與外部交聯劑之間的點擊反應是合成polyMOFs 的有效方法,但是這種方法難以實現聚合物鏈的規則排列。
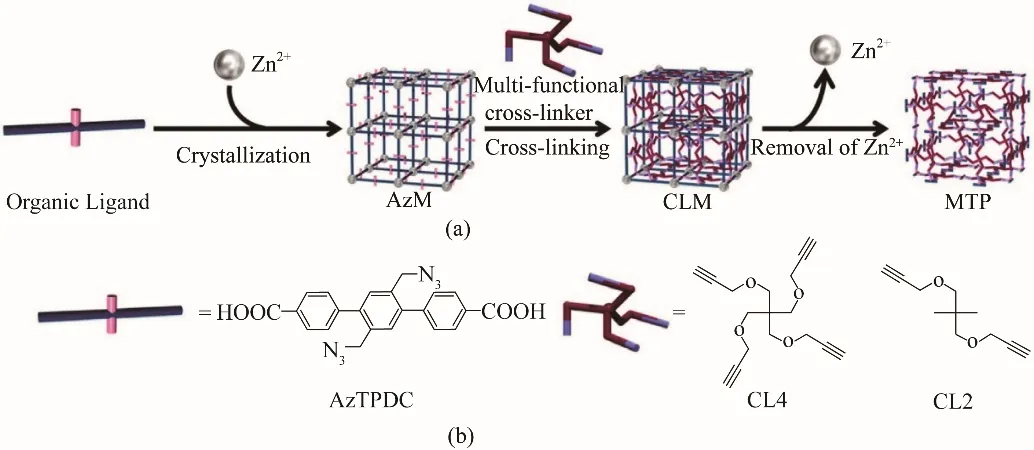
圖2 MOFs中有機配體的交聯和隨后分解以獲得聚合物凝膠(PG)的示意圖(a);有機配體和炔烴交聯劑的分子結構(b)[52]Fig.2 Schematic illustration of cross?linking of the organic linkers in MOF (AzM)and subsequent decomposition to obtain polymer gel(PG)(a).Molecular structures of the organic ligand(AzTPDC)and the cross?linkers(b) [52]
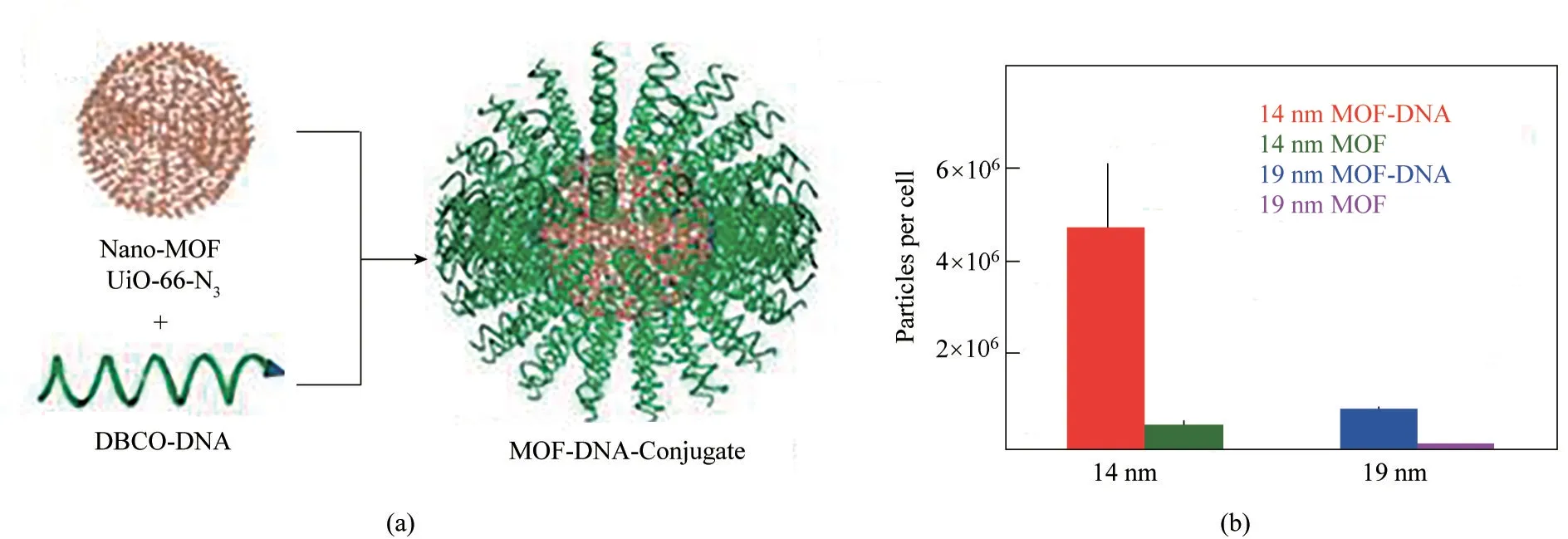
圖3 使用二芐基環辛炔(DBCO)官能化的DNA對UiO?66?N3納米顆粒進行功能化修飾(a);ICP?MS測定的細胞對納米顆粒的攝取量(b)[33]Fig.3 DNA functionalization of UiO?66?N3 nanoparticles,using DNA functionalized with dibenzylcyclooctyne(DBCO)(a).Nanoparticle uptake per cell determined by ICP?MS(b)[33]
Morris 等[33]利用點擊化學反應制備了MOFs 與DNA 相 互 偶 聯 的polyMOF 材 料(MOF?DNA?Conjugate)。他們先合成了疊氮基團官能化的MOF顆粒UiO?66?N3[Zr6O4OH4(C8H3O4?N3)6],然后利用二芐基環辛炔功能化的DNA 與UiO?66?N3之間的點擊反應制備了MOF?DNA?Conjugate,實現了寡核苷酸對MOF 表面的共價功能化。與未官能化的MOF顆粒相比,polyMOFs 的穩定性更高,細胞對polyMOFs 的攝取能力也更強(圖3),以孔徑為14 nm的MOFs 和MOF?DNA?Conjugate 為例,實驗細胞對前者的攝取量為3×105,對后者的攝取量為5×106。
1.2 MOFs孔道內的聚合
MOFs 孔道內的聚合即把小分子單體或者基團置于多孔材料的孔道內使其在孔道內發生聚合反應[47]。與點擊化學不同,這種在限域空間內進行的聚合反應往往能夠合成結構受控、分子量分布窄的聚合物,因而MOFs 孔道內的聚合對于控制聚合物的 立 構 規 整 度[54?56]、共 聚 物 序 列[57?58]和 自 組 裝 結構[59?60]等具有重要意義。
Furukawa 等[61]使用富含羥基的γ?環糊精(γ?CD)與K+合成了不同尺寸的環糊精MOF(CD?MOF),然后用乙二醇二縮水甘油醚將CD?MOF 晶體內部交聯,證明了MOFs 的配體可以在孔內交聯形成聚合物網絡。交聯前后的CD?MOF 具有明顯不同的性質,交聯前的CD?MOF 易溶于水,而交聯后得到的交聯CD?MOF不溶于水,熱穩定性也有所提升。
上述研究是利用配體上的羥基與交聯劑反應實現孔道內聚合,相較而言,使用乙烯基單體實現MOFs 孔道內的聚合研究更為廣泛,孔道內乙烯基單體的聚合往往能制備出聚合物鏈規整排列的polyMOF 材料。Distefano 等[62]將2,5?二乙烯基對苯二甲酸酯(DVTP)作為取代配體嵌入到多孔配位聚合物(PCP)的結構中,DVTP 可以同時連接PCP 上的金屬離子和PCP 孔道中的聚苯乙烯,將相鄰孔道的聚合物鏈橋接在一起,然后用Na2?EDTA 溶液選擇性溶解掉部分PCP 框架,實現了聚合物鏈高度有序的晶體堆積。Mochizuki 等[63]用單乙烯基官能化的配體X(苯乙烯3,5?二羧酸)作為有機配體合成MOFs(圖4),將苯乙烯基周期性地固定在MOF 框架中,另一種游離的乙烯基單體Y 滲入到MOFs 孔道后引發自由基聚合,制得框架上衍生單體X 和孔道中滲透單體Y 交替排列的polyMOF 材料。迄今為止,通過在MOFs 孔道內引入多種乙烯基聚合物,包括聚苯乙 烯[36,60,64?66]、聚 甲 基 丙 烯 酸 甲 酯[54]、聚 醋 酸 乙 烯酯[54]、聚丙烯腈[59]和聚(N?乙烯基咔唑)[67?68]等,已經合成多種polyMOF材料。
1.3 表面接枝
將聚合物接枝到MOFs表面是改善MOF材料性能的常用方法,目前最常見的表面接枝技術有“嫁接支鏈”(graft to)和“長出支鏈”(graft from)兩種[69?71]。“嫁接支鏈”,即預先合成的聚合物末端被官能化處理,帶有能夠與待接枝表面發生反應的基團,然后通過基團間的相互反應把聚合物接枝到MOFs 表面,這種接枝方法的缺點是接枝密度低,限制了聚合物的質量分數,不利于MOFs 的表面改性。與“嫁接支鏈”不同,“長出支鏈”是指先用引發劑將表面官能化,然后加入單體,單體在表面直接聚合長出聚合物鏈,這種方法能夠生長出高密度的聚合物,提升MOFs 表面的聚合物質量分數,有效改善MOFs的機械性與穩定性[72?74],因而“長出支鏈”法能夠合成性能較佳的polyMOF材料,所以本節重點介紹“長出支鏈”技術合成的polyMOF材料。
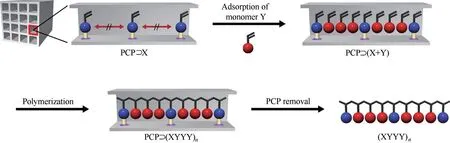
圖4 使用PCP進行序列調控的自由基聚合示意圖[63]Fig.4 Schematic illustration of sequence?regulated radical copolymerization using PCPs[63]
Xie等[35]通過從UiO?66?NH2表面生長聚乙二醇甲基丙烯酸甲酯(PEGMA)合成了水分散性極佳的polyMOF 材料。他們首先合成了UiO?66?NH2,然后使用原子轉移自由基聚合(ATRP)引發劑溴代異丁酰溴(BiBB)將MOF 顆粒表面功能化得到Br@MOF,之后在電子轉移再生催化劑ARGET?ATRP 的作用下從MOFs 表面生長出PEGMA 得到polyMOF 材料——P@MOF。如圖5 所示,將相同量的UiO?66?NH2與P@MOF 分散于水中,可以發現UiO?66?NH2顆粒在水中聚集且無法用超聲分散(左),而P@MOF由于聚合物的存在能夠以小顆粒的形式均勻分散在水中(右),具有良好的水分散性。將該polyMOF材料運用于將4?硝基苯酚(NP)還原為4?氨基苯酚(AP)的反應中。與Pd(0)負載的UiO?66?NH2相比,Pd(0)負載的P@MOF 具有更高的轉換頻率(TOF)和轉換數(TON),表現出了良好的催化活性。
早期開展的聚合物與MOFs 接枝研究往往會造成MOF 顆粒孔道堵塞和比表面積降低等。為了克服此問題,McDonald 等[75]設計了一種核?殼結構的polyMOF 材料,外殼既提供了聚合物生長的反應位點,又能很好地防止孔道堵塞。他們以MOF?5 為核,IRMOF?3 為殼,在MOF?5 的表面生長含有2?氨基對苯二甲酸酯連接基的IRMOF?3,然后利用IRMOF?3 上的胺基與2?溴異丁酸酐選擇性反應將引發劑安裝到IRMOF?3 的外殼上,在引發劑作用下,單體發生聚合形成了聚甲基丙烯酸甲酯(PMMA)。由于引發劑只位于殼上,所以聚合反應被限制在晶體的外殼中,MOF?5 核心完整無損(圖6)。此方法接枝PMMA 的MOFs 的表面積>2800 m2/g,與引發劑官能化后的MOFs 相比,表面積僅下降了15%。這表明核?殼結構能夠有效降低接枝技術中引起的孔堵塞問題,為合成高孔隙率的polyMOF 材料提供了一種思路。
近期Molavi 等[37]發現使用PMMA 接枝改性的UiO?66 對氦氣(He)具有良好的選擇滲透性。他們使用甲基丙烯酸縮水甘油酯(GMA)將UiO?66?NH2官能化而獲得乙烯基附著的UiO?66 (GMA?UiO?66),之后MMA 單體原位聚合,直接從GMA?UiO?66顆粒表面生長出PMMA,在MOF 顆粒周圍形成了聚合物殼,得到了polyMOF材料(PMMA?g?GMA?UiO?66)。然后以此polyMOF 材料為分散相,以PMMA 為連續相制備了混合基質膜。接枝的PMMA 增加了MOF 顆粒的分散性與穩定性,改善了PMMA 基質與MOF 納米顆粒之間的界面相互作用,使得混合基質膜中MOF 摻雜量可高達28%,在He/CH4及He/N2的混合氣測試中,該混合基質膜對He表現出較高的通量及選擇性。此外,通過力學性能表征發現純PMMA 膜的楊氏模量為1.7 GPa 左右,而摻雜polyMOFs 后混合基質膜的楊氏模量最高可達2.7 GPa,表明polyMOFs 的添加能夠明顯改善混合基質膜的力學性能。
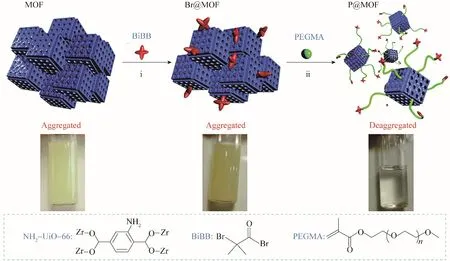
圖5 制備P@MOF的示意圖[35]Fig.5 Schematic representation of the preparation of P@MOF[35]
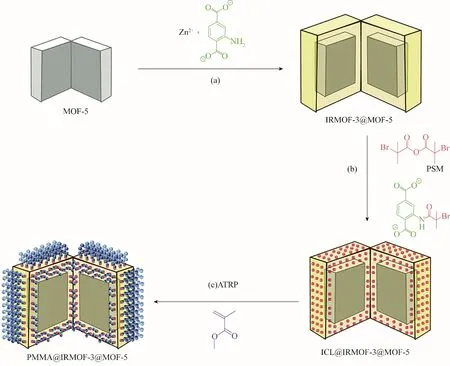
圖6 PMMA@IRMOF?3@MOF?5的合成路線[75]Fig.6 Synthetic route to PMMA@IRMOF?3@MOF?5[75]
“長出支鏈”一般指的是從MOFs 顆粒表面接枝聚合物,而Saleem 等[32]制備了一種從聚合物表面長出MOF 晶體的polyMOF 材料,并將其應用于糖基化肽的識別。他們通過自由基聚合合成苯乙烯?二乙烯基苯?甲基丙烯酸三元共聚物(terpolymer),然后利用層層自組裝技術在聚合物表面長出了ZIF?8,之后用氨基苯硼酸進行官能化處理,合成了terpolymer@ZIF?8@BA。此外,還測試了該polyMOF材料對免疫球蛋白G?單糖基化和HRP 多糖基化的富集情況。結果表明,聚合物的引入增強了MOF 材料的親水性與再生性,能夠特異性識別并高效富集糖蛋白和糖肽,具有從復雜生物樣品中提取糖基化肽的潛力,可用于輔助基于HRP 的蛋白質組學分析。這種從聚合物表面長出MOFs 晶體的技術對于推動polyMOFs材料的多元化發展具有重要意義。
2 單晶到單晶轉換
單 晶 到 單 晶(single?crystal to single?crystal,SCSC)轉換是指反應前的原料與反應后的產物均為晶體,但是反應過程中可能涉及到材料結構的變化,比如配位模式、材料構型的改變[76]。由于將有機聚合物直接摻入MOFs 比較困難,因此可以通過SCSC 轉換間接合成polyMOF 雜化材料。SCSC 轉換可以在溶劑或非溶劑環境下進行,其中固態SCSC反應是在無溶液的環境下進行的,可以排除溶液等其他因素的影響,是制備polyMOF 材料最直接有效的方法[77?78]。引發SCSC 轉換的外界刺激源有很多,常見的有溫度變化、紫外線照射、溶劑蒸氣的吸收或揮發、機械作用等,目前主要通過溫度變化和光化學反應引發的SCSC轉換來制備polyMOF材料。
2.1 溫度變化引發的SCSC轉換
溫度是影響MOFs 結構的重要因素,通過加熱去除溶劑分子可以誘導晶體發生固態SCSC 轉換。溫度的變化甚至可能引起整個晶體框架的改變,因而可以通過控制溫度誘導低維聚合物轉變為三維立體的polyMOF材料。
L?ssig等[79]通過加熱將一維配位聚合物[Cd(Me?3py?trz?pba)2(H2O)2]·H2O(Me=甲基,py=吡啶,trz=連三唑,pba=苯甲酸)轉化為三維結構。當加熱溫度為230 ~290 ℃時,一維聚合物失去配位水分子,Cd2+配位結構改變形成了三維結構的polyMOF 材料(圖7)。這種通過溫度變化制備的polyMOF材料是在熱力學推動下形成的,因而具有極高的熱穩定性,這為制備熱穩定性良好的polyMOF材料提供了一種思路。
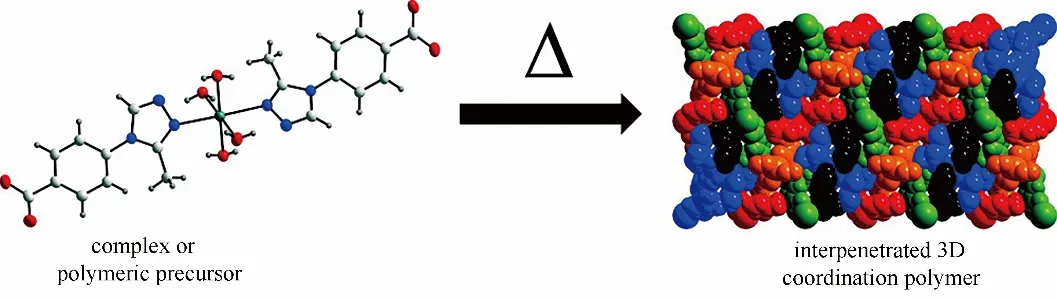
圖7 加熱作用下一維配位聚合物轉化為三維立體結構示意圖[79]Fig.7 Schematic diagram for the transformation from 1D coordination polymer chains to 3D frameworks by heating[79]
2.2 光化學引發的SCSC轉換
相比于溫度變化引發的SCSC 轉換,光化學引發的SCSC轉換研究更深入。光化學引發SCSC轉換是指僅通過外界光源照射即可實現晶體內分子的重排或轉化,而不需要額外的環境條件。與溫度變化引起的SCSC 轉換不同,光化學反應過程通常不涉及溶劑分子的損失,而是利用雙鍵的環加成反應將框架彼此連接起來,制備polyMOF材料。同時,利用光化學進行SCSC 轉換合成polyMOF 材料的過程中往往伴隨著維度的變化,例如,通過紫外線誘導的[2+2]環加成反應將低維度的材料轉化成三維MOF結構[80?83]。
2012 年,Ou 等[80]首次報道了光化學反應引發的1D 鏈到3D 框架的SCSC 轉換。他們使用不同的金屬離子與1,2?雙(4'?吡啶基)乙烯(bpe)和反式?1,2?環己烷二甲酸酯(1,2?chdc)反應制得一維鏈狀結構的[M(1,2?chdc)(bpe)2(H2O)2]·H2O (M=Zn 或Mn)。其中,1,2?chdc 配體上的兩個羧酸酯基團以單齒模式連接兩個鋅原子,充當形成一維Zn(1,2?chdc)鏈的“橋”,bpe 配體向四個方向延伸,在汞燈照射下相鄰的bpe 配體發生[2+2]環加成反應生成了3D 結構的polyMOF材料(圖8)。
Medishetty 等[81]利用兩種含有4?苯乙烯基吡啶(4?spy)的配位聚合物層層堆疊,進行[2+2]環加成光聚合反應,伴隨著單晶至單晶結構轉變,2D 交錯層轉變為具有互穿網絡結構的3D MOFs。此后,他們進一步研究了其他含有C====C 的吡啶基配體MOFs 2D 至3D 的 轉 變[82]。Xie 等[83]利 用[Mn2L2(H2O)2]·3H2O(H2L = E?5?(2?(吡啶?4?基)乙烯基)間苯二甲酸)配體也實現了2D 至3D 的紫外線誘導的[2+2]環加成反應。
上述研究都是先形成配位聚合物然后通過環加成制備三維的polyMOF 材料,因而會伴隨著維度的變化,這種合成過程是不可逆的,制備的polyMOF材料不能夠回到低維度的聚合前狀態。而Park等[84]證實了polyMOF 材料的可逆合成,他們利用1,4?雙[2?(4'?吡啶基)乙烯基]苯(bpeb)配體中C====C 雙鍵的環加成反應在MOFs 中合成聚合物,成功將包含環丁烷環的有機聚合物引入MOFs,把具有六重互穿網絡結構的原始MOF[Zn(bpeb)(bdc)]n轉換為具有新網絡結構的[Zn(poly?bppcb)(bdc)]n(poly?bppcb = 1,3?(4,4'?聯吡啶)?2?苯基環丁烷聚合物),同時保留了Zn2+周圍的四面體結構。加熱可將環丁烷環裂解,再次獲得原始結構,證明這兩種結構之間的轉變是可逆的。并在此基礎上通過[2+2]環加成反應將MOF 轉化為由有機配體和二維配位聚合物組成的金屬?有機聚合物框架(MOPF)[85]。
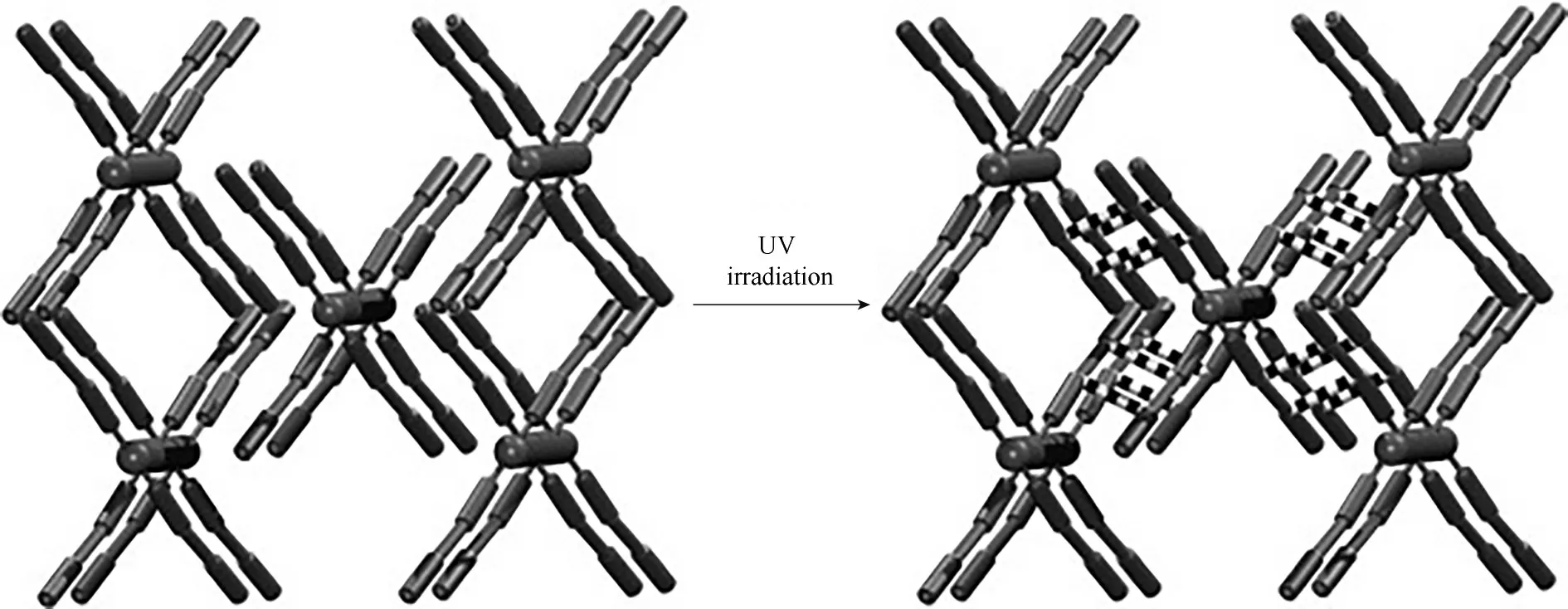
圖8 通過[2+2]光二聚作用從1D交錯式鏈到3D框架的轉換示意圖[80]Fig.8 Schematic diagram for the transformation from 1D staggered?sculls chains to 3D frameworks through[2+2]photodimerization[80]
在上述研究的基礎上,Park 等[86]還通過SCSC 轉換在柱狀MOF 中可逆地引入了間同立構聚合物。原始MOF [Zn2(bpeb)(obc)2]n[obc = 4,4'?氧雙(苯甲酸乙酸酯)]在不同的溶劑中的光反應性不同,當孔道中的DMF或甲醇被水代替時,bpeb配體在空間上的位置排布滿足光聚合條件,通過環加成反應,生成了同時包含二維配位聚合物和有機聚合物配體的新型MOF,該MOF還表現出了光致發光特性以及對有機小分子的敏感性。Park 等是在MOF 框架形成后通過紫外線誘導的[2+2]環加成反應將聚合物引入MOFs,這種方式制備的polyMOFs 中的聚合物往往可以在加熱后解聚,還能夠形成不同構象的聚合物,這對功能性polyMOF 材料的設計與合成具有重要意義。
3 直接合成法
polyMOFs 的直接合成是指利用配位化學誘導線性、非晶態聚合物形成三維多孔的晶體結構,此過程中聚合物直接與各種金屬離子(例如Zn2+、Zr4+等)相互作用折疊成高度結晶的polyMOF 材料。與間接合成法相比,直接合成采用一鍋合成法,合成步驟簡單。但是目前關于直接合成法制備polyMOF材料的研究還處于初始階段,這些研究主要集中在MOFs 框架結構與聚合物鏈段(如間隔基團的長度、同構網狀擴張[87]和嵌段的共聚物)的關系上。
Zhang 等[88]首次制備出了IRMOF?1 型結構的polyMOFs,實現了由一維線性、無孔、無定形聚合物合成三維、多孔、結晶polyMOF 材料的目標(圖9)。他們使用Williamson 醚合成法制備了主鏈含有對苯二甲酸單元(H2bdc)的一維線性聚合物配體(pbdc?xa),H2bdc 單元之間的間距可以通過亞甲基的個數(即x值)來調節。實驗結果表明,當亞甲基個數為5~8 時可以制備出高結晶度的材料,并且與IRMOF?1具有相同的晶體結構。此外,將IRMOF?1 與polyIRMOF?1 暴露在空氣中一段時間,通過PXRD檢測晶體峰值的變化,結果表明與IRMOF?1 相比,使用聚合物配體制備出的polyIRMOF?1 具有良好的穩定性,這是因為聚合物配體中的烷基鏈增加了MOF材料的疏水性。
確定一維線性聚合物配體能夠成功合成MOF框架后,Zhang 等[89]進一步合成了多種晶體結構的polyMOF 框架,比如同時使用聚合物配體和吡啶基配體[如1,2?雙(4?吡啶基)乙烷(bpe)、4,4'?聯吡啶(bpy)等]合成了柱狀polyMOFs 并詳細對比了相應的MOFs與polyMOFs的水穩定性與氣體吸附性能。結果顯示,Zn?pbdc?12a(bpy),Zn?pbdc?11a(bpe)和Zn?pbdc?12a(bpe)這三種polyMOF 材料在測試條件下能保持其結晶度和原始形貌,表明polyMOF 材料具有極佳的穩定性。此外,氣體吸附數據顯示,這些polyMOF 材料對CO2的吸附量最高可達(140±5)cm3/g,而幾乎不吸附N2,具有分離煙道氣中CO2/N2的潛力。
上述polyMOFs 的配體都是通過Williamson 醚合成法制備的,其分子量低且分布不均勻。Ayala等[90]用無環二烯復分解法(ADMET)制備了分子量較高且分布均勻的聚合物配體,并使用該配體合成了基 于Zr4+的UiO?66 型polyMOFs。實 驗 發 現,當H2bdc 單元之間的間距適當時,polyUiO?66 MOFs 具有交錯形態的層狀介孔結構,這表明聚合物配體能夠用于生成具有分層結構的MOF 材料,這是小分子配體合成的母體UiO?66材料所不具備的特性。
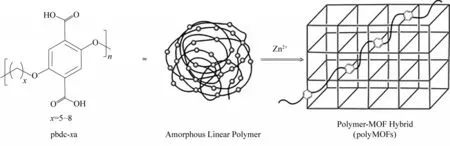
圖9 將一維(線性)、無孔、大部分為非晶態的聚合物轉變為三維、多孔、結晶polyMOF雜化材料的策略[88]Fig.9 The strategy described herein to convert a one?dimensional(linear),non?porous,mostly amorphous polymer into a three?dimensional,porous,crystalline polyMOF hybrid material[88]
Cohen 等[87,91?92]以及Yazaki 等[93]進一步探討了聚合物配體的結構對polyMOFs 的影響。Schukraft等[87]利用同構網狀化學法(isoreticular chemistry)制備了包含1,4?聯苯二羧酸(H2bpdc)和1,4?三聯苯二羧酸(H2tpdc)單元的軸向擴展的聚合物配體,并研究了這種同構網狀擴展得到的聚合物配體對系列polyUiO?MOFs 和polyIRMOFs 的影響。結果表明,與常規聚合物配體制備的polyMOFs相比,軸向擴展的聚合物配體制備出的polyMOF 材料表面積更大。以系列polyUiO?MOFs為例,BET測試結果顯示常規聚合物配體制備的polyUiO?66 比表面積為(414±8)m2/g,而軸向擴展的聚合物配體制備的polyUiO?67?8a?u,polyUiO?68?8a?u 和polyUiO?68?10a?u 的 比表面積分別是(618 ± 180) m2/g、(1626 ± 190) m2/g、(1342 ± 100) m2/g,同時穩定性也有所提升。Cohen等[94]又研究了聚合物配體上醚氧鍵的位置(對位取代=p?pbdc、鄰位取代=o?pbdc)對polyMOFs 的影響( 圖10),發 現 使 用o?pbdc 的 配 體 能 夠 合 成polyIRMOF?1但是不能合成基于Zr4+的polyUiO?66。Yazaki 等[93]通過陽離子聚合反應制備了對苯二甲酸基(bdc)位于側鏈的聚合物配體,由此側鏈型聚合物配體合成了polyMOF?5 材料。上述研究均證實了聚合物配體的結構具有影響或限制polyMOFs 形成的作用。
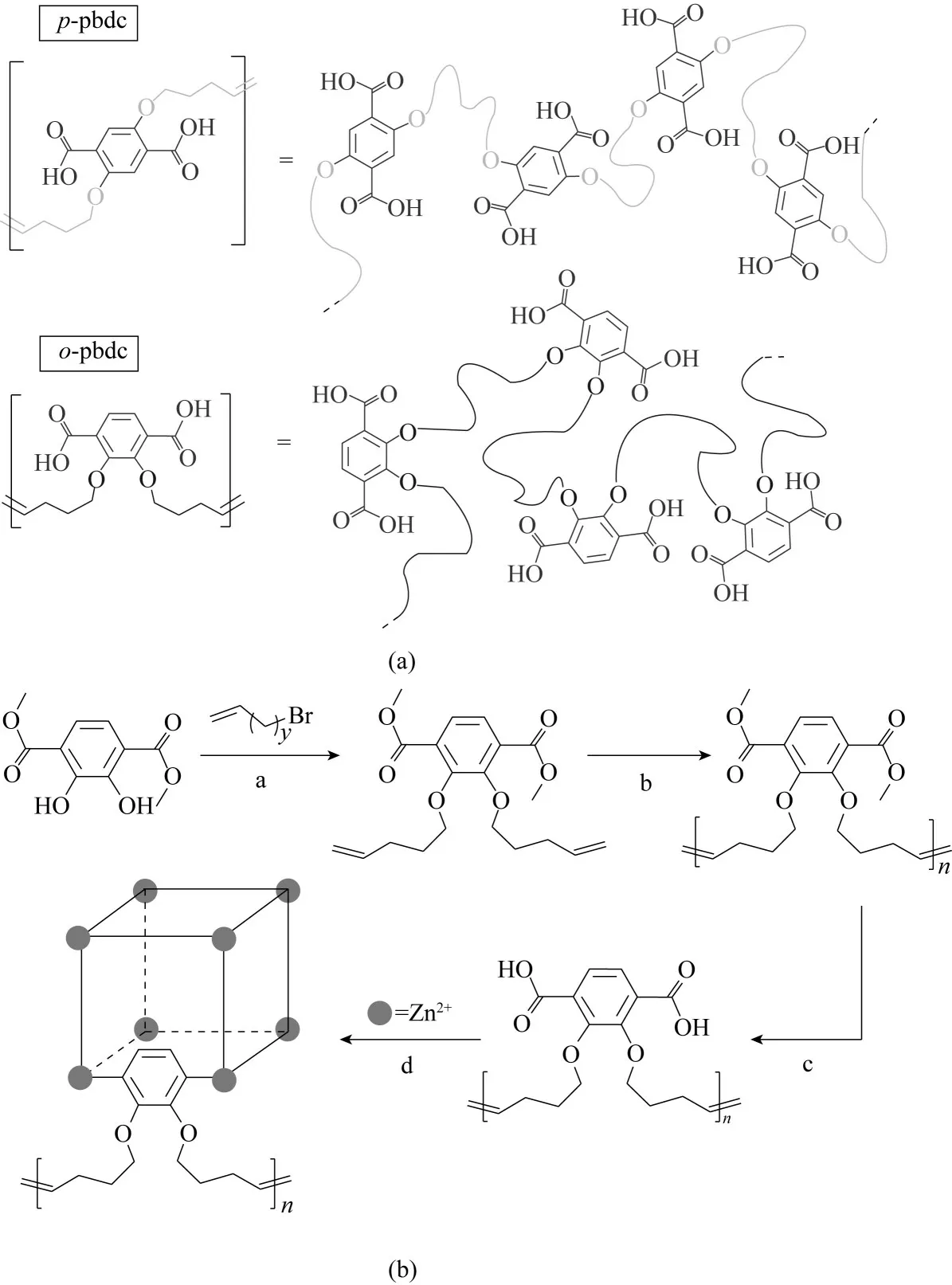
圖10 對位取代(上)和鄰位取代(下)聚合物配體的比較(a);o?pbdc?xa?u配體的制備及polyIRMOF?1的合成(b)[91]Fig.10 Comparison of para?substituted(top)and ortho?substituted(bottom)polymer ligands(a).Synthesis of o?pbdc?xa?u and subsequent formation of polyIRMOF?1(b)[91]
嵌段共聚物(BCP)可以整合多種聚合物的優勢,具有分子量可控且分子量分布較窄等特點,而且嵌段共聚物具有在溶液中自發組裝形成更高階組裝體的能力[94],所得組裝體的尺寸和形貌在很大程度上取決于嵌段共聚物的總分子量和每個嵌段的質量分數[95?96],這些特性使得嵌段共聚物在制備功能性polyMOFs 方面有很好的前景。Macleod 等[97]首次使用嵌段共聚物合成了嵌段共聚polyMOFs(block co?polyMOFs,BCPMOFs)。所用BCP由兩部分組成,其中一個嵌段是通過迭代指數增長(IEG)合成的H2bdc 二聚體或三聚體,另一個嵌段是通過原子轉移自由基聚合制備的聚苯乙烯(PS),這兩段通過銅點擊化學偶聯,然后與Zn2+作用制備BCPMOF 材料。實驗發現H2bdc 二聚體形成的聚合物配體不能夠生成晶體狀的polyMOF 材料,這表明H2bdc 單元的數量會影響BCPMOFs的形成。此外,他們發現由嵌段聚合物配體制備的polyIRMOF?1 存在MOF 組分和PS 組分之間的納米相分離,表明這些BCPMOF 材料具有類似于嵌段共聚物的性能。在此基礎上,Gu等[98]以H2bdc 和PEG 的嵌段共聚物配體作為大分子調節劑同時實現了MOF 納米顆粒的尺寸控制和表面功能化,可控地合成了直徑數十至數百納米的polyMOF?5 納 米 顆 粒(polyMOF?5 NP)。普 通 的MOF?5 對水蒸氣耐受性很差,在空氣中放置數小時后會分解成MOF?69c[99],而Gu 等發現polyMOF?5 在空氣中放置6周后依然能夠保持原有結構。由于表面存在不飽和配位點和未配位的配體,MOF?5 納米顆粒往往會相互反應發生聚沉,而polyMOF?5 NP表面被PEG 包裹,能夠有效防止納米顆粒的聚集,大大提升了納米顆粒的膠體穩定性。
而后,Cohen 等[100]研究了嵌段共聚物的組成(嵌段的結構和質量分數)對polyMOFs 形貌的影響。他們使用了兩種嵌段,H2bdc 嵌段用于配位形成BCPMOFs,PEG 或聚環辛二烯(polyCOD)嵌段用于調控BCPMOFs的形貌,兩種嵌段可以在溶液中自發組裝。研究發現,通過改變PEG或polyCOD的分子量、質量分數等能夠明顯影響BCPMOFs 的形貌(圖11),進而會對材料的晶體結構、比表面積、孔徑、孔隙率等產生影響。Cohen 等的工作證明了嵌段共聚物能夠影響polyMOFs 的形貌,這為polyMOFs 的可控合成提供了一種方便簡單的思路,極大地推動了polyMOFs的發展。
Gao 等[38]提出了一種通過原位合成的聚合物接枝MOF 來消除混合基質膜中的界面缺陷的方法。使用Zn2+、2?甲基咪唑和超支化PEI,在室溫下原位合成了polyMOF 材料——PEI?g?ZIF?8,如圖12 所示。在PEI?g?ZIF?8 中,PEI 上的胺基參與金屬離子的配位,將聚合物嵌入MOF 架構中,有效地改善了MOF 框架與聚合物的相容性,與ZIF?8 相比,PEI?g?ZIF?8 具有更高的孔體積、更大的比表面積,穩定性也得到了提升。以PEI?g?ZIF?8材料為分散相,聚乙烯胺(PVAm)為連續相制備混合基質膜(PVAm/PEI?g?ZIF?8/PSf)。由于引入了PEI,PEI?g?ZIF?8 材料與聚合物具有良好的界面相容性。如表1所示,與PVAm/ZIF?8/PSf膜[101]相比,相同條件下制備的PVAm/PEI?g?ZIF?8/PSf 膜中PEI?g?ZIF?8 顆粒的最佳摻雜量高達20%,且CO2滲透速率提升了167%。
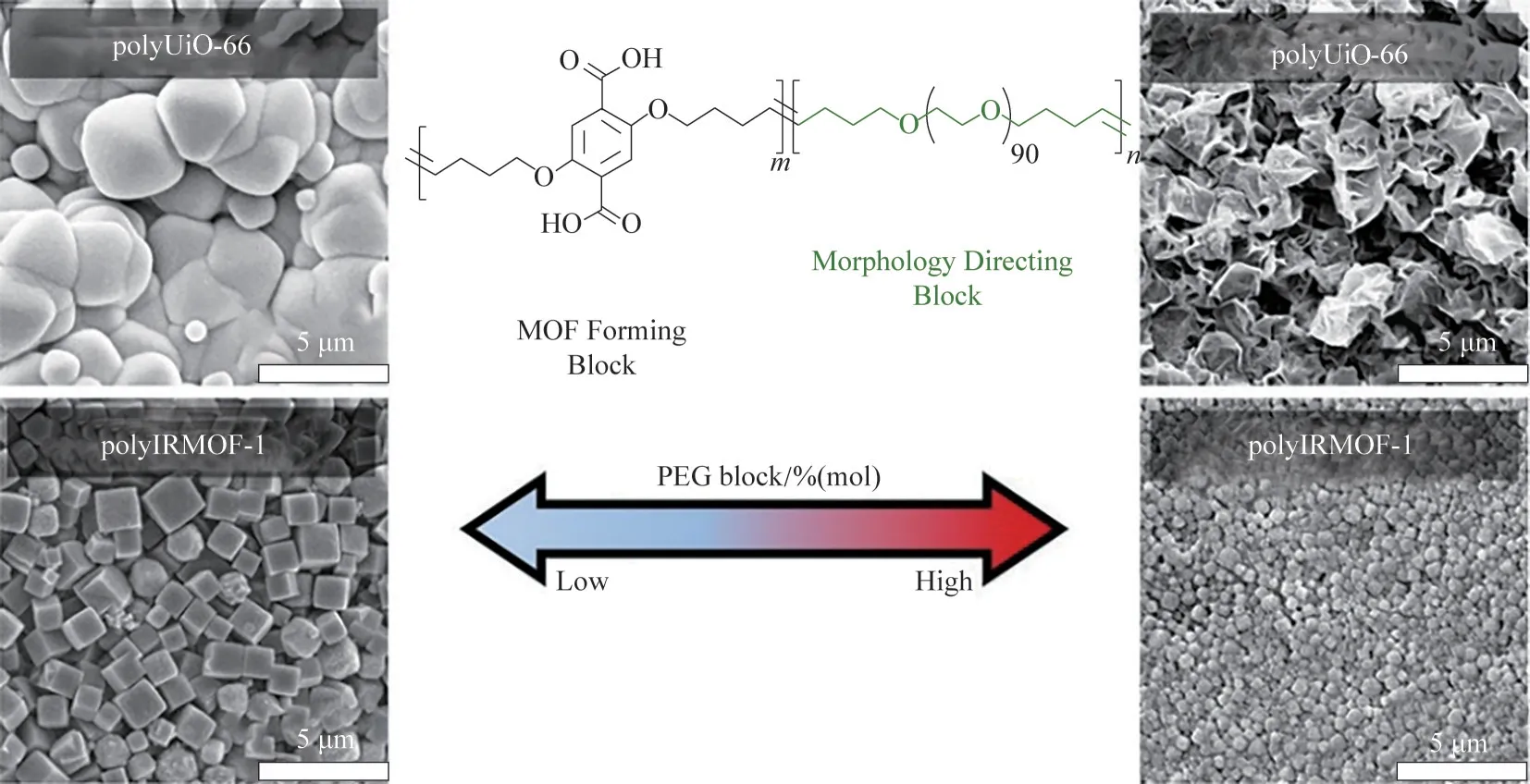
圖11 嵌段共聚物的結構和質量分數對polyMOFs形貌的影響[100]Fig.11 Effect of block copolymer architecture and mass fractions on polyMOFs morphology[100]
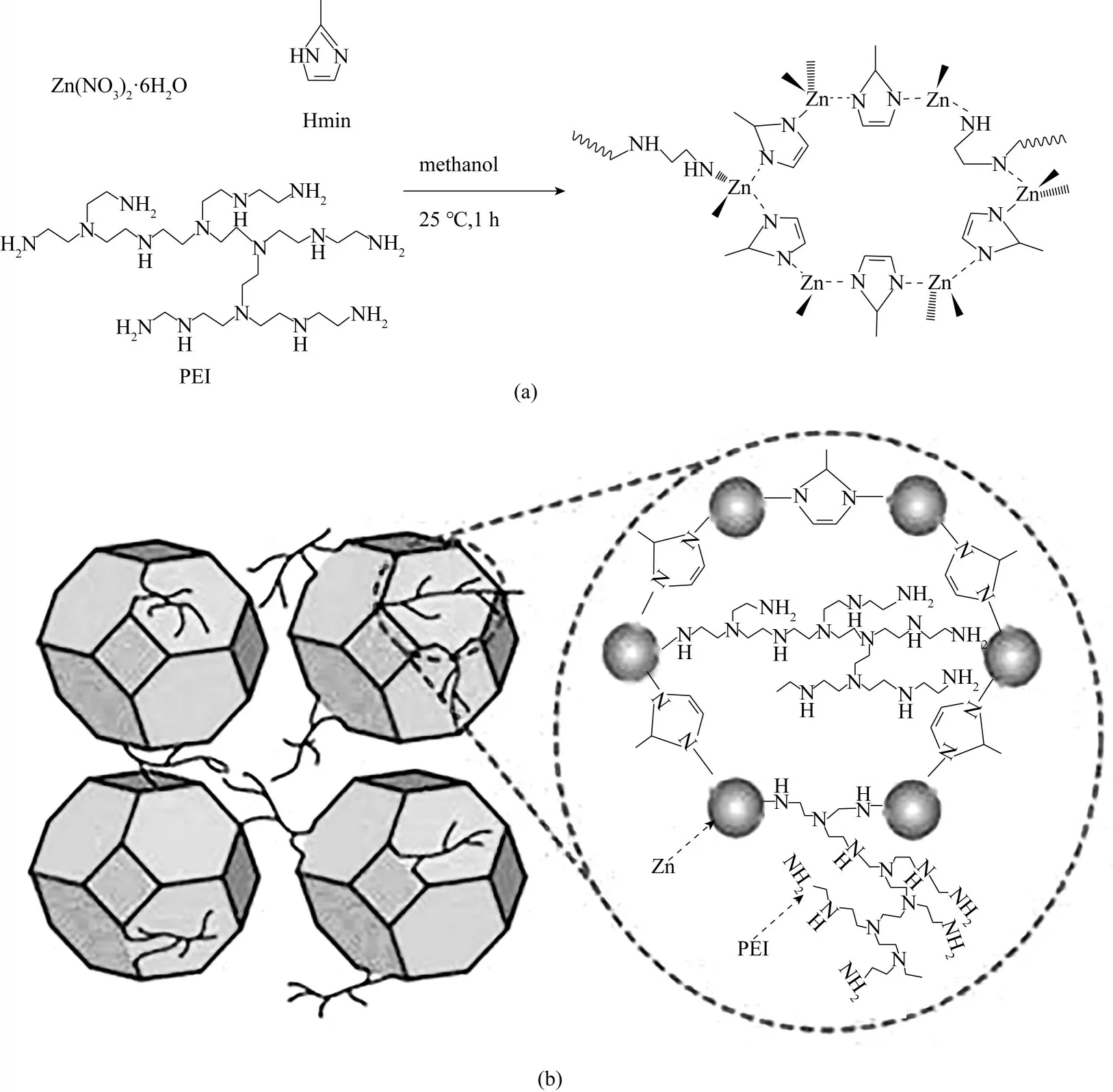
圖12 PEI?g?ZIF?8納米顆粒的原位合成方案(a);PEI?g?ZIF?8納米顆粒的結構圖(僅顯示了Hmim和PEI的部分主要連接體)(b)[38]Fig.12 In situ synthesis protocol of PEI?g?ZIF?8 nanoparticles(a).Structure illustration of PEI?g?ZIF?8 nanoparticles(only showing some main linkers of Hmim and PEI)(b)[38]
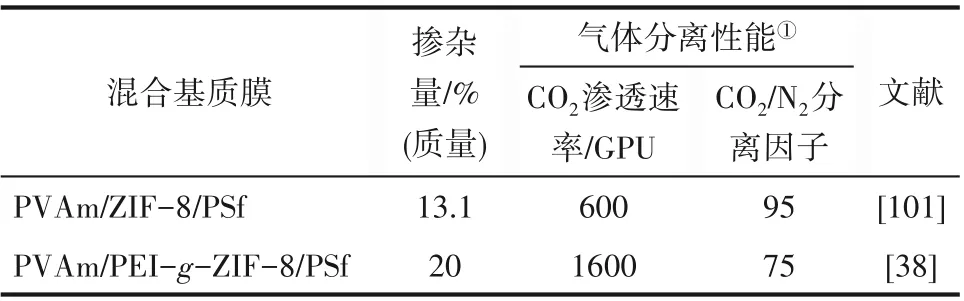
表1 兩種混合基質膜的摻雜量及氣體分離性能對比Table 1 Comparison of doping amount and gas separation performance of two mixed matrix membranes
事實證明,與原始MOFs 相比,將聚合物作配體的一部分所合成的polyMOFs 具有更高的穩定性[89]、更優異的可加工性[97]與功能性[38]。但是,上述案例中涉及的polyMOFs 均是以MOFs 為模板,是在已有MOFs 的基礎上制備的,并沒有從根本上改變其原有的晶體結構。Qiao 等[39?40]以聚合物為模板,開創性地利用聚合物作為polyMOFs 的框架的支柱(pillar),通過直接合成法制備出了具有新型晶體結構的微孔聚合物。這種微孔聚合物具有大比表面積、規整有序的貫通孔結構和尺寸可調的孔徑,可以作為氣體快速傳遞通道應用于氣體分離膜的制備。
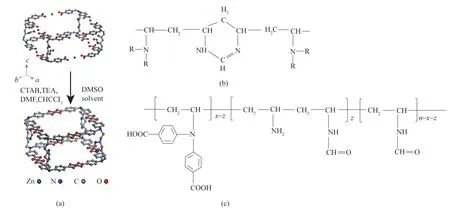
圖13 MPF?1晶體的主要框架結構(a);MPF?1晶體主要框架中的聚合物鏈段(b);PVAmacid的結構(c)[39]Fig.13 The main structure of each independent MPF?1 framework(a).The polymer segments of the main MPF?1 framework(b).The structure of PVAmacid(c) [39]
首先,Qiao 等[39]合成了一種有序微孔聚合物——金屬誘導的聚合物框架1(MPF?1)[(圖13)]。在DMSO 溶劑中,聚合物配體PVAmacid[結構如圖13(c)]與Zn2+配位連接形成平均孔徑為0.94 nm 的MPF?1 晶體[圖13(a)]。MPF?1 具有兩大優勢,可用于制備性能良好的CO2分離膜。①MPF?1是具有一定柔韌性的高分子量聚合物,因此具有良好的成膜能力;②MPF?1具有均勻的納米級孔徑(0.94 nm),并含有可與CO2分子發生可逆反應的胺基,因此,CO2分子可以優先吸附在MPF?1 的孔壁上,并通過單分子表面擴散跨孔運輸,而大多數N2、CH4或H2分子則被排除在孔外。Qiao 等利用10%(質量)MPF?1水分散液制備了30 μm 濕涂層厚度的MPF?1/PSf 膜,以CO2/N2(體積比為15/85)、CO2/CH4(體積比為10/90)和CO2/H2(體積比為40/60)混合氣作為原料氣測試膜的氣體分離性能,在323 K,0.5 MPa下膜對三種混合氣的CO2滲透速率均為1050 GPU 左右,CO2/N2分離因子為51,CO2/CH4分離因子為81,CO2/H2分離因子為41。
而后,Qiao 等[40]采用聚合物定向化學合成法(PDCS)合成了金屬誘導的微孔聚合物,稱為MMPs。以金屬離子Cu2+、Zn2+作為配位中心,以線性高分子聚合物(PEI、PVAm)作為框架支柱(pillar),在合成過程中,小分子有機連接基(醋氯芬酸、4?氯間苯二甲酸)首先與Cu2+或Zn2+配位,然后與不同分子量的PVAm 或者PEI 連接,通過自組裝過程將單元結構與聚合物鏈有序連接來構建MMPs 框架。合成方法、框架單元結構及晶體結構如圖14 所示。PVAm 骨架和晶體尺寸的可控性可以避免缺陷的出現,進一步提高了膜的分離性能。將MMPs 水分散液涂覆在聚乙烯醇(PVA)和聚二甲基硅氧烷(PDMS)改性的PSf(mPSf)基膜上,MMPs 以豎直、非堆疊態在基膜表面緊密排布,從而形成超薄且無缺陷的氣體分離膜。該膜有規整的高滲透性氣體傳輸通道,因此具有良好的CO2分離性能。以MMP?3 膜為例,其CO2滲透速率可達3000 GPU,CO2/N2(體積比15/85)分離因子可達78,且分離性能在0.2 MPa 以內保持不變。
在此基礎上,Yuan等[102]采用改進的PDCS工藝,合成了一種新型高價金屬誘導的有序多孔聚合物HMMP?1。與MMPs 相比,HMMP?1 具有強金屬?氧配位鍵和高密度的游離胺基,并在強堿性環境中具有良好的化學穩定性。以HMMP?1 為分散相,PVAm 為連續相制備混合基質膜。由于聚合物框架與連續相PVAm 良好的界面相容性,HMMP?1 納米顆粒的最佳負載量可達44.4 %(質量),此時該膜在0.5 MPa下的CO2滲透速率為1200 GPU,CO2/N2(體積比15/85)分離因子為150。
與通過直接混合聚合物和納米材料制備的MMMs 不同,MPF?1、MMPs 和HMMP?1 的每個晶體單元均包含聚合物鏈段,因此,它們具有良好的界面相容性并顯示出優異的CO2分離性能。這種PDCS 策略也可以應用于其他聚合物,從而有可能實現微孔聚合物的簡單合成,制備新型polyMOF 雜化材料。
4 總結與對比
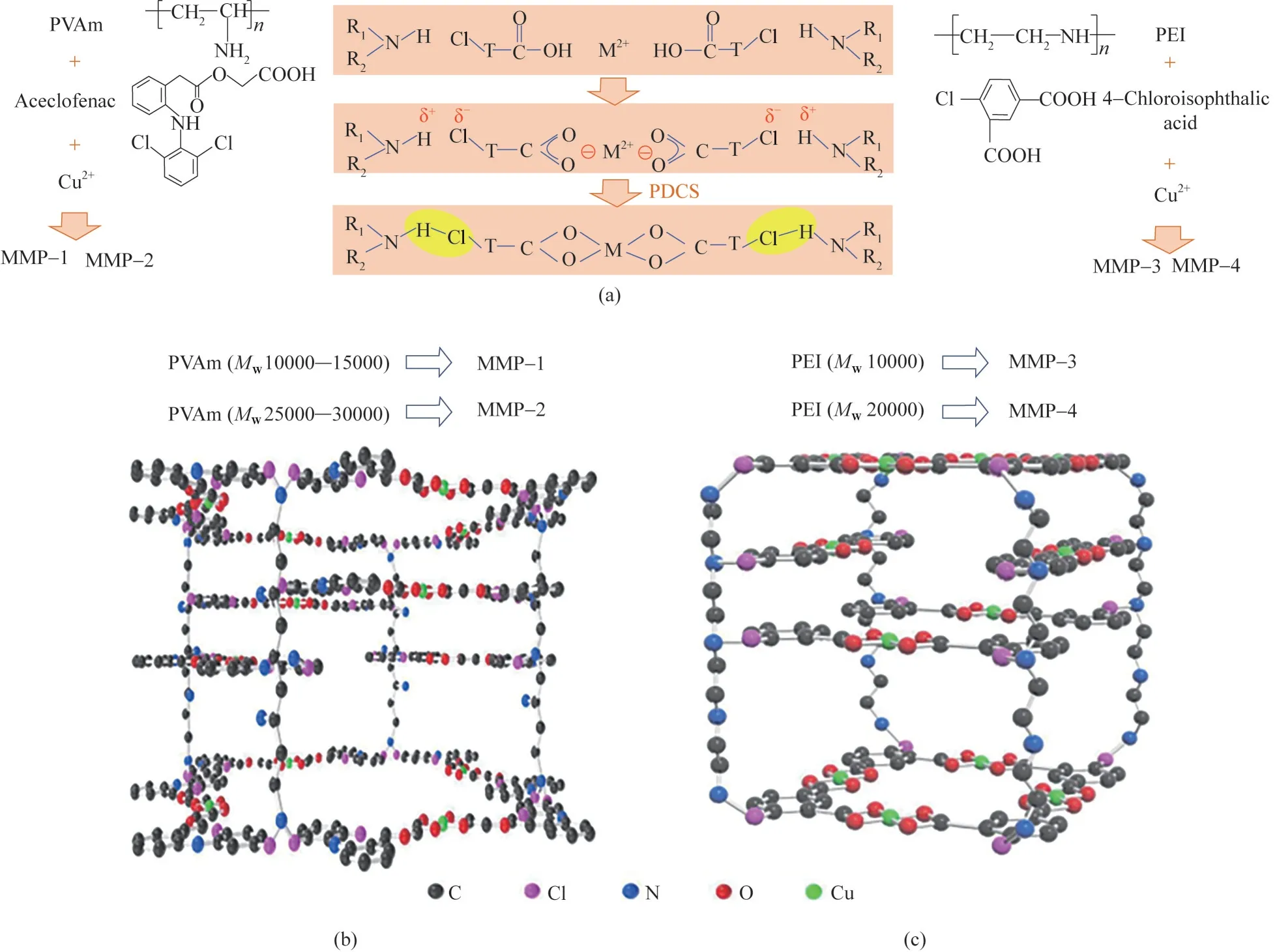
圖14 使用PDCS構建MMP框架(a);MMP?1和MMP?2的框架結構(b);MMP?3和MMP?4的框架結構(c)[40]Fig.14 Construction of MMP frameworks using the PDCS process(a).Independent frameworks of MMP?1 and MMP?2(b).Independent frameworks of MMP?3 and MMP?4(c)[40]
polyMOFs 的出現突破了材料軟與硬、結晶性與非晶性之間的界線,開拓了聚合物與MOFs 雜化研究的新領域。polyMOFs整合了聚合物與MOFs的優勢,具備高氣體吸附選擇性、良好的水解穩定性、良好的生物相容性和易加工性等特點,這些優勢通過單一的聚合物或者MOFs 都難以實現,使其在催化、生物醫學、氣體分離等領域具有廣闊的應用前景。本文介紹了polyMOF 材料的三種合成方法:合成后聚合、單晶到單晶轉換和直接合成法,在此總結了不同方法合成的典型polyMOF 材料,并對比了不同合成方法的優缺點,如表2 所示。可以看出每種合成方法的優劣各不相同。由于材料設計及制備過程相對簡單,科研人員對表面接枝法的研究較多,其應用也最為廣泛;光化學引發的SCSC 轉換,能夠可逆地合成polyMOF 材料,但是其熱穩定性一般較差,這極大地限制了其應用范圍;直接合成法制備出的polyMOF 材料整體性能往往優于另外兩種方法,是近幾年研究的熱點。總體來說,在制備polyMOF 材料時,應根據需求選擇合適的合成方法。
5 展 望
盡管polyMOF 材料具有諸多優勢,但是其合成與應用仍存在許多挑戰。例如,聚合物的引入在增強MOFs 性能的同時極易造成孔道堵塞,降低材料的孔隙率,覆蓋部分反應位點,嚴重阻礙了polyMOF材料的應用。此外,在結構表征、反應機理探究和孔徑調控等方面仍需要更深入的研究。因此,未來關于polyMOF 材料的研究可能會沿以下幾個方面開展:
(1)深入研究polyMOFs 的晶體結構。大部分文獻中的polyMOFs 結構都是通過表征與計算模擬推測出的,具有一定的不確定性,需要對晶體結構式、原子配位形式等進行更深入的研究,提供更直觀、更具說服力的晶體結構。
(2)探究polyMOFs 的合成機理。目前關于polyMOFs 的合成仍處于探索階段,很少涉及機理方面的研究,如配位機理、溶劑效應、結構調控等[2,7,103],合成機理研究的匱乏導致功能性polyMOF材料的設計與合成難度較大,重復性也較差,限制了polyMOF 材料的進一步應用,因此研究者們應對反應機理進行更深入的探索,尤其是反應動力學及反應熱力學方面。

表2 不同合成方法制備的典型polyMOF材料及其優缺點Table 2 Typical polyMOFs synthesized by different methods and their advantages and disadvantages
(3)精準調控polyMOFs 的孔徑。目前報道的論文中幾乎沒有關于polyMOFs孔徑調控的研究,聚合物鏈段的引入會對MOFs 材料的孔徑造成較大的影響,導致合成的polyMOF 材料與初始的母體MOFs相比,孔結構和比表面積都發生改變,聚合物鏈段的折疊和相互堆積使得polyMOFs 的孔徑調控變得十分復雜,而孔徑的精準調控具有十分重要的意義,尤其在氣體分離領域,孔徑的大小與分布會直接影響到材料的氣體分離性能。因而如何有效調控polyMOFs 材料的孔徑大小與孔徑分布關系到其應用前景。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
