SLC12A3基因突變致Gitelman綜合征一家系報道并文獻復習*
2021-03-17 05:54:46朱智峰閆朝麗
現代醫藥衛生 2021年5期
朱智峰,閆朝麗
(內蒙古醫科大學附屬醫院內分泌科,內蒙古 呼和浩特 010050)
Gitelman綜合征是腎臟遠曲小管離子轉運蛋白先天缺陷的代表性疾病,也是遺傳性腎小管疾病中患病率最高的,與Bartter綜合征同屬于失鹽性腎病。1966年,GITELMAN等[1]首次描述了其是一種不同于Bartter綜合征的遺傳性失鹽性腎病,主要表現為低血鉀、低血鎂、低尿鈣、代謝性堿中毒、腎素-血管緊張素-醛固酮系統活化、正常血壓,其主要與編碼腎臟遠曲小管鈉-氯協同轉運蛋白的SLC12A3基因突變有關,在臨床中很容易被漏診。本文對3例同一家系Gitelman綜合征患者臨床表現、診斷方法、治療與轉歸進行總結分析,并參閱、復習國內外相關文獻。
1 臨床資料
患者1,女,35歲,因“間斷四肢抽搐12年”于2016年8月入本院。患者12年前自覺情緒激動后間斷出現四肢及口周麻木、抽搐感,伴有心悸及心前區憋悶感,一般持續10 min左右可自信緩解,未予重視。2016年7月無明顯誘因再次出現上訴癥狀,持續約10 min左右自行緩解,癥狀較之前無明顯加重,就診于包頭醫學院第一附屬醫院,急查血離子:血鉀2.33 mmol/L,血鈣2.62 mmol/L,血鎂0.43 mmol/L;24 h尿鉀121 mmol/L。血氣分析:pH 7.46,碳酸氫根31.3 mmol/L,血鉀2.5 mmol/L,血鈣1.07 mmol/L。腎上腺CT薄層掃描:左側腎上腺內中段類圓形脂肪密度影,腎上腺髓質瘤?給予對癥補鉀及補鎂(具體劑量不詳)治療后,血鉀及血鎂仍持續低水平波動。為進一步明確診治就診于本院,門診以“低血鉀原因待查”收入院。病程中,無四肢乏力,無肢體癱瘓,無呼吸困難,無口干,多飲、多尿,無惡心、嘔吐,無腹痛、腹瀉,飲食及睡眠可,喜食咸食,大小便正常,近半年體重無明顯下降。無特殊既往史。查體:血壓120/80 mm Hg(1 mm Hg=0.133 kPa),身高160 cm,體重50 kg,體重指數(BMI)19.5 kg/m2,甲狀腺不大,心、肺、腹查體未見異常。脊柱呈正常生理彎曲,四肢運動自如,雙下肢無水腫,生理反射存在,病理反射未引出。
患者2,女,28歲,因“間斷雙手抽搐6年余,加重2年,發現血鉀低1周”于2016年8月入本院。患者6年前無明顯間斷出現雙手抽搐,1年發作2次左右,每次持續約3~5 min可自行緩解,未在意。2年前上訴癥狀加重,間斷時間縮短,每次持續時間較前延長(具體不詳),伴有嘴角抽搐,心前區不適,無全身乏力,未重視,未診治。1周前體檢發現血鉀低(2.89 mmol/L),為進一步診治就診于本院,門診以“低鉀血癥”收入院。病程中,患者無心悸、氣短,無惡心、嘔吐,無腹痛、腹瀉,飲食及睡眠可,喜食咸食,大小便正常。近半年體重無明顯下降。無特殊既往史。查體:血壓120/80 mm Hg,身高160 cm,體重55 kg,BMI 21.5 kg/m2,甲狀腺不大,心、肺、腹查體未見異常。脊柱呈正常生理彎曲,四肢運動自如,雙下肢無水腫,生理反射存在,病理反射未引出。
患者3,女,24歲,因“間斷四肢乏力伴發作性麻木抽搐6個月,加重2 d”于2017年10月入本院。患者6月前無明顯誘因出現四肢乏力,未在意,后出現四肢麻木抽搐,雙手為著,伴雙上肢發僵、屈曲受限。就診于北京市昌平市中醫院化驗血鉀2.4 mmol/L,給予靜脈補鉀治療后癥狀緩解(具體補鉀量不詳)。復查血鉀2.5 mmol/L,患者拒絕進一步輸液補鉀。后間斷口服氯化鉀緩釋片,未復查血鉀變化。上述癥狀先后發生6次,最近2次間隔10 d,最近一次于入院前2 d發生,再次就診于北京市昌平區中醫院。化驗血鉀3.0 mmol/L,給予靜脈補鉀3.0 g,復查血鉀3.9 mmol/L,為系統診治入本院治療。病程中,無呼吸困難,無口干,多飲、多尿,無惡心、嘔吐,無腹痛、腹瀉,飲食及睡眠可,喜食咸食,大小便正常,近半年體重無明顯下降。無特殊既往史。查體:血壓101/70 mm Hg,身高155 cm,體重48 kg,BMI 20 kg/m2,甲狀腺不大,心、肺、腹查體未見異常。脊柱呈正常生理彎曲,四肢運動自如,雙下肢無水腫,生理反射存在,病理反射未引出。
3例患者為同一家系,其家族史:父母體健,非近親結婚。
1.2方法
1.2.1檢測方法 電解質利用ISE-900離子檢測儀采取離子選擇電極法檢測,血氣分析利用Radiometer ABL 90系列檢測,腎素-血管緊張素-醛固酮系統利用Autolumo A2000全自動化學發光測定儀采用化學發光法檢測,皮質醇利用Beckman Coulter Unicel Dxi800全自動免疫分析儀進行免疫化學法檢測,口服葡萄糖耐量試驗、肝腎功能采用貝克曼AU5831全自動生化分析儀檢測,甲狀腺功能利用Cabase 601分析儀采取化學發光法檢測,采用免疫熒光法檢測抗中性粒細胞胞漿抗體(ANCA)、可提取性核抗原(ENA),心電圖為十二導聯心電圖,雙腎采取超聲檢查,基因檢測送北京協和醫院檢測。
1.2.2檢測結果 3例患者甲狀腺功能、口服葡萄糖耐量試驗、腎臟超聲、心電圖及血、尿、便常規和肝、腎功能未見異常,ANCA、ENA、皮質醇、促腎上腺皮質激素水平。血清電解質水平、24 h尿電解質水平、血氣分析及醛固酮、腎素水平見表1~3。進一步行基因檢測,3例患者均存在SLC12A3基因突變,4號外顯子上有一個純合錯義突變:c.533C>T,178TCG>TTG,Ser178Leu;23號外顯子上有一個雜合錯義突變:c.2738G>A/G,913CGG>CAG,Arg913Gln。進一步對患者母親及父親進行基因檢測:母親基因正常,父親基因存在4號和23號外顯子雜合突變,但無臨床癥狀,且血鉀等檢查結果皆正常。見圖1、2。

表1 血清電解質水平(mmol/L)
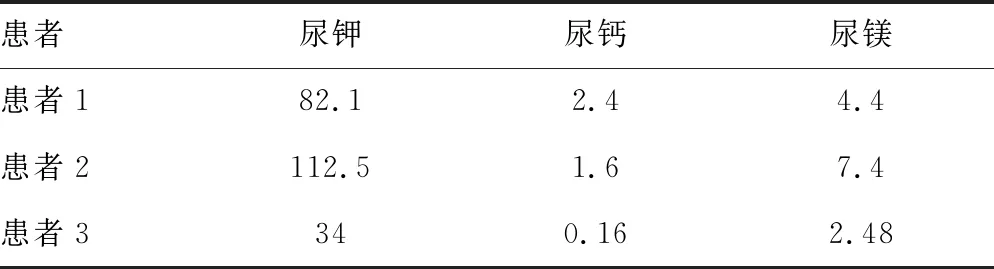
表2 24 h尿電解質水平(mmol/L)

表3 血氣分析及醛固酮、腎素水平
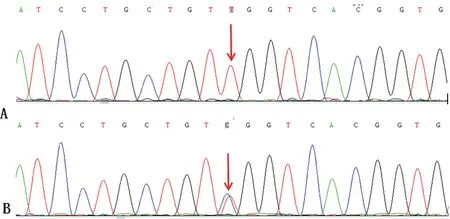
A為純合突變(患者);B為雜合突變(父親)
1.2.3治療方法 3例患者均給予口服氯化鉀緩釋片(每次2 g、每天3次)、門冬氨酸鉀鎂片(每次2片、每天3次)、螺內酯(每次20 mg、每天2次),癥狀好轉后出院。出院半年后隨訪,復查結果:患者1血鉀3.0 mmol/L,血鎂0.52 mmol/L;患者2血鉀3.03 mmol/L,血鎂0.57 mmol/L;患者3血鉀3.3 mmol/L,血鎂0.5 mmol/L。
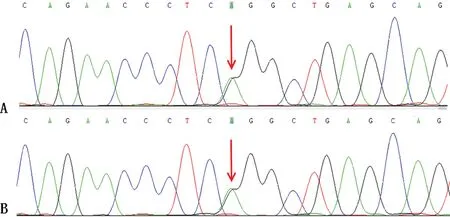
A為雜合突變(患者);B為雜合突變(父親)
2 討 論
Gitelman綜合征是一種罕見的常染色體隱性遺傳性疾病,其病變部位在腎臟遠曲小管。Gitelman綜合征病因是編碼位于腎臟遠曲小管的噻嗪類利尿劑敏感的鈉氯共同轉運體蛋白的基因SLC12A3發生功能缺失突變,導致其結構和(或)功能異常所致。目前,Gitelman綜合征確切發病率尚不清楚,歐洲約為1/40 000,亞洲可能更高。根據雜合子攜帶率估算,日本Gitelman綜合征發生率為10.3/10 000。Gitelman綜合征主要臨床表現為低血鉀、低血氯性堿中毒、低血鎂、低尿鈣、血壓正常或偏低,以及腎素-血管緊張素-醛固酮系統活化。典型Gitelman綜合征患者可通過臨床表現和實驗室檢查獲得臨床診斷,而最終確診則有賴于基因檢測[2]。改善全球腎病預后組織(KDIGO)于2017年Gitelman綜合征爭議會議共識中制定了詳細診斷標準[3]。Gitelman綜合征均以低鉀血癥為首發表現,且起病隱匿,因此以低鉀血癥的病因鑒別為切入點展開討論。
低鉀血癥的原因主要有:(1)攝入不足、胃腸道丟失或鉀離子異常分布的患者多數存在胃腸道疾病病史或周期性麻痹,尿鉀檢查提示無腎性失鉀;慢性嘔吐或腹瀉患者可存在低血鉀及尿鉀排泄增多,但其尿鉀排泄不增高(血鉀小于3.5 mmol/L,尿鉀小于25 mmol/d,或血鉀小于3.0 mmol/L,尿鉀小于20 mmol/d),無腎性失鉀;使用利尿劑患者可存在低血鉀、高尿鉀和失氯,需仔細詢問用藥史。(2)腎性失鉀,主要表現為低血鉀、高尿鉀(血鉀小于3.5 mmol/L,尿鉀大于25 mmol/d,或血鉀小于3.0 mmol/L,尿鉀大于20 mmol/d)。如果低血鉀患者合并高血壓,還需進行皮質醇等檢測并結合影像學檢查排除原發性醛固酮增多癥、Cushing綜合征、腎素瘤、腎動脈狹窄、Liddle綜合征等;血壓不高時,需要與一些自身免疫性病如干燥綜合征及某些藥物引起的腎小管損傷相鑒別,因為這些疾病均可出現Gitelman綜合征或Bartter綜合征樣表現,此時需要通過病史、臨床表現、自身抗體檢測、血氣分析等檢查加以鑒別。
3例患者為親姐妹,均為青年女性,血壓不高,均表現為嚴重低血鉀、高尿鉀,考慮為腎性失鉀,進一步行血氣分析檢查提示為代謝性堿中毒。3例患者血壓均不高,符合這樣特征的疾病主要有Bartter綜合征、Gitelman綜合征、干燥綜合征、長期嘔吐、藥物性(腎毒性藥物、氨基糖苷類藥物、利尿劑)等。通過詢問病史及體格檢查,3例患者均無長期嘔吐史及腎毒性藥物、氨基糖苷類藥物、利尿劑等藥物服用史,且無口干、眼干、猖獗齒表現,風濕免疫相關檢查為陰性,而且患者3人為親姐妹,高度懷疑為Bartter綜合征、Gitelman 綜合征。
Bartter綜合征是包括低血鉀、代謝性堿中毒、正常血壓、高醛固酮血癥和對血管緊張素Ⅱ反應減弱的一組癥候群,腎活檢提示腎小球旁器增生。Gitelman綜合征是一種不同于Bartter綜合征的常染色體隱性遺傳性失鹽性腎病,主要表現為低血鉀、低血鎂、低尿鈣、代謝性堿中毒、腎素-血管緊張素-醛固酮系統活化、正常血壓。1966年,SIMON等[4]通過基因連鎖分析確定Gitelman綜合征的突變基因是位于16號染色體上,由編碼腎臟遠曲小管噻嗪敏感的鈉氯協同轉運蛋白的SLC12A3基因突變使其功能失活所致。Gitelman綜合征診治專家共識[2]提到,Gitelman綜合征和Bartter綜合征均有低血鉀、代謝性堿中毒高腎素活性,但是Gitelman綜合征還有低血鎂、低尿鈣,發病時間主要在青少年或成年期,生長發育遲緩較少見,其病變部位主要為腎臟遠曲小管SCL12A3基因突變。而Bartter綜合征并無低血鎂、低尿鈣,其發病部位在腎臟髓袢升支粗段,因編碼NKCC2及相應的調節蛋白基因突變所致,對水、鹽的重吸收作用較腎臟遠曲小管更關鍵。因此,Bartter綜合征發病時間早,主要在兒童期,生長發育遲緩較多見,臨床癥狀更嚴重。3例患者為成年起病,除了有低血鉀、高尿鉀、代謝性堿中毒,同時存在低血鎂、低尿鈣、腎素-血管緊張素-醛固酮系統活化。進一步行基因檢測,在SLC12A3基因中發現2個致病突變。Gitelman綜合征的診斷需結合患者臨床癥狀和實驗室檢查,而且必須強調低鎂血癥和低尿鈣癥的重要性,如果缺乏這2個條件,一般不能診斷Gitelman綜合征[5]。目前,越來越多的文獻報道,少數Gitelman綜合征患者血鎂是正常的[6-7]。
Gitelman綜合征的發病機制:腎臟遠曲小管的功能障礙導致Na+和Cl-重吸收減少,水丟失過多,導致血容量減少,從而激活腎素-血管緊張素-醛固酮系統,導致低鉀血癥和代謝性堿中毒。低鎂血癥可能因為在腎小管頂端膜上存在Mg2+轉運通道TRPM6,在基底膜上通過Mg2+/Na+交換增加,而使尿鎂增加,血鎂降低[8]。目前,血鎂正常的Gitelman綜合征患者也越來越多地被確診。國內外文獻報道,Gitelman綜合征中正常血鎂患者比例為8%~22%[9]。北京協和醫院腎內科研究團隊在Gitelman綜合征患者腎活檢組織中觀察到,遠端腎小管上Mg2+轉運蛋白TRPM6與鈉氯協同轉運蛋白共表達,正常血鎂患者TRPM6與健康人表達接近,而低鎂患者TRPM6表達顯著降低;低血鎂患者較正常血鎂患者臨床表現及鈉氯協同轉運蛋白功能損害嚴重,從而為正常血鎂亞型的確立提供了病理生理基礎[9-10]。尿鈣降低可能原因是,管腔側Na+重吸收減少,則基底膜側Na+/Ca2+交換增加,因而管腔側Ca2+重吸收增加,尿鈣減少。
Gitelman綜合征的臨床表現,主要為低血鉀及低血鎂所導致的骨骼肌、心血管、胃腸道、腎臟、神經系統的表現。Gitelman綜合征基因突變情況,目前已發現近400種SLC12A3的基因突變,錯義突變最常見,其中僅18%為純合突變,45%以上為復合雜合突變,且大于7%的患者擁有3個或3個以上的突變位點,但突變位點的多少與臨床表現無相關性[11]。國外不同地區的突變位點不同,歐洲人以IVS9+1G>T多見[12],我國則以T60M及D486N突變多,推測Gitelman綜合征人群的發生率為1/40 000~10/40 000,且亞洲人群患病率可能更高[13]。2018年北京協和醫院腎內科完成140例患者的基因檢測,其中105例發現基因突變而確診為Gitelman綜合征,共發現69種突變[14]。在國內文獻報道的249個中國人家系的125種突變(476個受累等位基因)中,攜帶T60M和D486N的比例分別為14.1%和8.4%,是中國人群Gitelman綜合征的高頻突變,且國內報告病例數較多的單位是北京協和醫院、上海瑞金醫院、青島醫學院附屬醫院[14-16]。本研究中,3例患者SLC12A3基因突變位點均為Ser178Leu和Arg913Gln,不是高頻突變。3姐妹均為4號外顯子純合突變、23號外顯子雜合突變,其父親4號外顯子和23號外顯子都是雜合突變,但母親是正常的。可能突變的4號外顯子等位基因是患者自身突變導致了純合致病,但也有可能是患者母親會存在一個片段缺失情況,剛好遺傳給了患者。患者從父親那里接受了雜合突變,又從母親那里接受了片段缺失,因此,直接測序測到純合突變,但需要進一步性定量聚合酶鏈反應檢測大片段來驗證。
Gitelman綜合征的治療目標主要是改善患者癥狀,提高患者生活質量,同時避免嚴重并發癥。Gitelman綜合征的治療原則主要是不限制鈉鹽的攝入,多進食富含鉀離子的食物,同時給予補鉀、補鎂和針對其病理生理機制的利尿劑治療,其中補鉀及補鎂為重要的治療部分。KDIGO建議血鉀及血鎂的治療目標分別為3.0 mmol/L和0.6 mmol/L[3]。本研究中,3例患者經過口服補鉀、補鎂、螺內酯治療,肢體及抽搐癥狀較前明顯好轉。但是,停止用藥后癥狀會再次加重,如患者2停用藥物后出現突發心悸、四肢無力,心電圖顯示V1~V6導聯T波,考慮為心肌缺血,經住院繼續口服補鉀、補鎂、螺內酯及短期靜脈補充鉀鎂,癥狀好轉,血鉀及血鎂水平進一步提高。
綜上所述,臨床上遇到低血鉀、高尿鉀、低血鎂、低尿鈣、代謝性堿中毒且血壓不高的患者時,應考慮Gitelman綜合征可能,但其確診依賴于基因檢測,且盡量對其家系進行基因測序以明確診斷。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
