
液相色譜串聯質譜法測定蜂蜜中咪唑類抗生素
2021-04-01 04:22:50吳明徐飛
食品工業 2021年3期
關鍵詞:檢測
吳明,徐飛
1. 寧夏回族自治區食品檢測研究院(銀川 750002);2. 寧夏回族自治區疾病預防控制中心理化科(銀川 750004)
硝基咪唑類(Nitroimidazoles)和苯并咪唑類(Benzimidazoles)抗生素是一類用于動物寄生蟲治療的驅蟲劑,其中硝基咪唑類藥物由于低廉的價格,被廣泛用于蜜蜂孢子蟲病的預防和治療[1]。試驗證明,蜂蜜中殘留的硝基咪唑類和苯并咪唑類抗生素具有明顯的致畸、致癌和致突變作用。因此,包括我國在內的許多國家和地區明確規定,動物源食品中咪唑類和苯并咪唑類抗生素不得檢出或限定了最大使用量[1-2]。隨著生活水平的日益提高,國家監管機構進一步加大對咪唑類藥物殘留問題的監督,為保證相關法律法規的實施,需要較高的技術水平和儀器設備對蜂蜜中的獸藥殘留進行檢測。具有高檢測精度、高靈敏度和高通量等特點的高效液相色譜-串聯質譜(LC-MS/MS)法成為檢測蜂蜜中咪唑類抗生素的主要技術之一[3-7]。由于蜂蜜樣品基質中存在大量的蛋白質、脂類和酚類等雜質干擾測定結果的可靠性,且咪唑類藥物在多處于痕量水平,因此,在儀器分析之前,必須采用固相萃取法等前處理方法除去雜質干擾和基質效應對結果的影響[3-7]。在前處理方面,采用多次推送過濾凈化(m-PFC)法萃取和凈化。m-PFC操作簡單,只需吸取提取液快速數次通過固相吸附劑,即可在數十秒達到凈化目的。m-PFC被廣泛用于不同樣品基質中農藥和獸藥的前處理工作[8-10]。在色譜分離方面,硝基咪唑類抗生素在普通C18反相色譜柱上易出現難以保留,以及苯并咪唑類抗生素在C18反相色譜柱峰形差和檢出限偏高等問題。試驗采用m-PFC凈化,分別選用強陽離子交換柱和五氟苯基柱分離分析硝基咪唑類抗生素和苯并咪唑類抗生素,正離子模式下檢測蜂蜜中殘留的藥物取得良好試驗效果。m-PFC-LC-MS/MS法操作快速簡單、重現性好,可用于蜂蜜中硝基咪唑類和苯并咪唑類抗生素的檢測。
1 材料與方法
1.1 儀器和試劑
LC-20AD UFLC(日本SHIMADZU);4000 Q-Trap LC-MS/MS(美國AB SCIEX);3-30K臺式冷凍離心機(瑞士SIGMA);FMC-1000恒溫振蕩器(日本EYELA);Milli-Q超純水儀(美國MILLIPORE);B-400均質儀(瑞士BUCHI)和漩渦混合器(德國IKA)。
16種硝基咪唑類和苯并咪唑類抗生素標準品(天津阿爾塔,1ST9241-100M,溶劑為甲醇,規格100 μg/mL)。
Ultimate NX-SCX柱(2.1 mm×150 mm,3 μm)和Ultimate NX-PFP柱(2.1 mm×100 mm,3 μm)(上海月旭科技股份有限公司);Quick Pro HF-S型凈化柱(北京華仁健康科技有限公司,IC-QuE3150-HF-S,150 mg/3 mL);甲酸(色譜純,美國ACS);乙腈為色譜純(德國Merck);其他試劑均為國產優級純。
2018—2019年間采集的30份蜂蜜均為寧夏省風險監測項目抽檢樣品,采樣過程符合國家標準要求,保存于4 ℃冰箱中,備用。
1.2 液相色譜-串聯質譜法
1.2.1 硝基咪唑類抗生素的色譜條件
色譜柱采用Ultimate NX-SCX柱(2.1 mm×150mm,3 μm);柱溫40 ℃;流速0.3 mL/min;進樣體積10.0 μL;流動相A為0.1%甲酸水,B為乙腈;梯度洗脫程序:0.0~0.5 min,10% B;0.5~2.0 min:10%~90% B;2.0~4.0 min 90% B;4.0~5.0 min,90%~10% B;5.0~7.0 min,10% B。
1.2.2 苯并咪唑類抗生素的色譜條件
Ultimate NX-PFP柱(2.1 mm×100 mm,3 μm);柱溫40 ℃;流速0.3 mL/min;進樣體積10 μL;流動相A為0.1%甲酸水,B為乙腈;梯度洗脫程序:0.0~0.5 min,10% B;0.5~2.0 min:10%~90% B;2.0~6.0min,90% B;6.0~7.0 min,90%~10% B;7.0~10.0 min,10% B。
1.2.3 質譜條件
離子源采用電噴霧離子源(ESI);掃描方式采用正離子掃描(ESI+);檢測方式采用多反應監測(MRM);氣簾氣(CUR)20 psi;電噴霧電壓(IS)5 500 V;離子源溫度(TEM)550 ℃;霧化氣(GS1)40 psi;輔助氣(GS2)40 psi。16種硝基咪唑類和苯并咪唑類抗生素的相關質譜參數見表1。
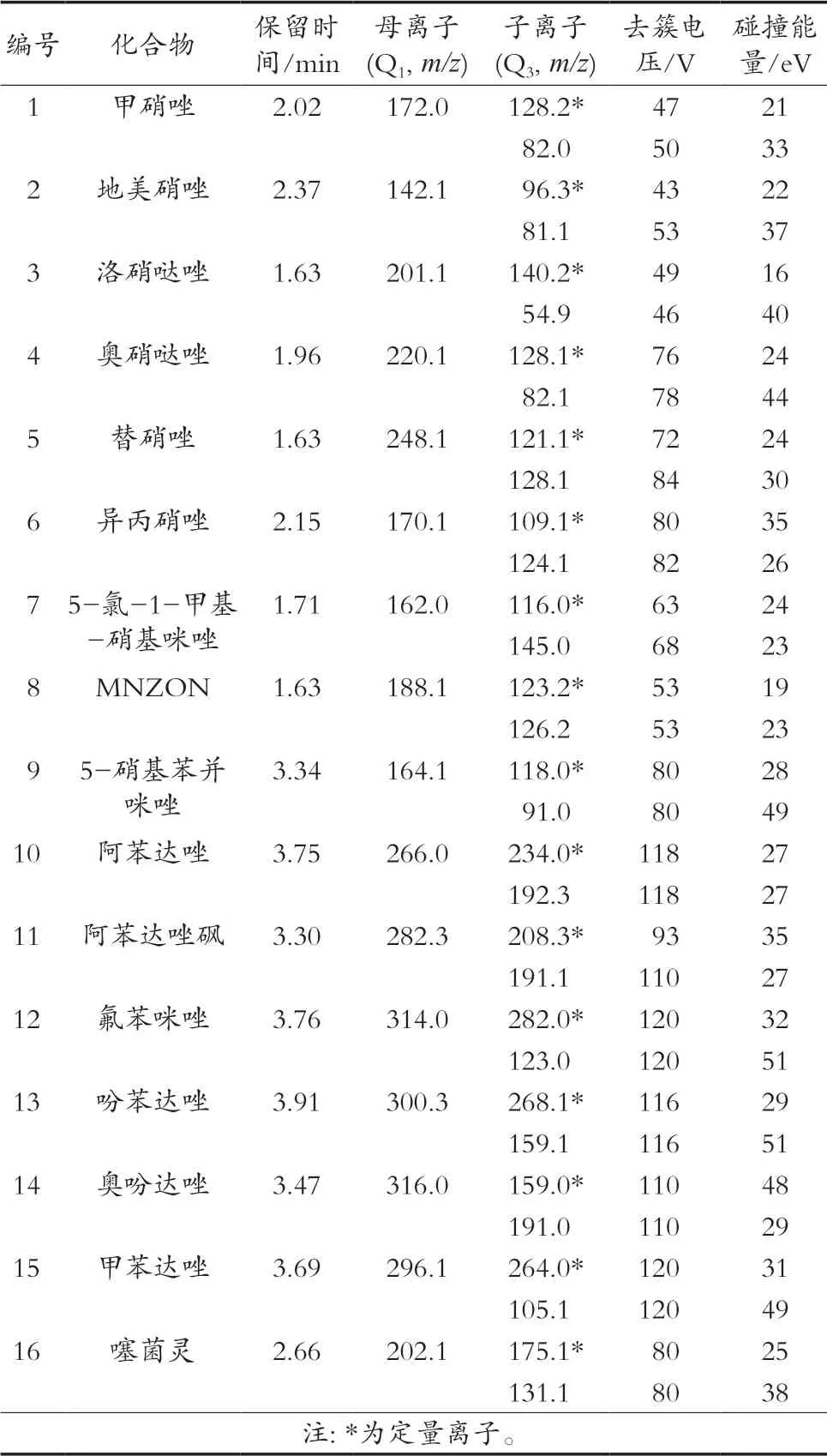
表1 質譜采集參數
1.3 提取凈化
1.3.1 提取
準確稱取5.0 g蜂蜜樣品于50 mL離心管中,加入2.0 g無水硫酸鎂,加入10 mL乙酸乙酯,渦旋混勻30s,4 ℃下以7 500 r/min離心5 min,收集上層乙酸乙酯于另一離心管中,加入10 mL乙酸乙酯,重復上述步驟,合并乙酸乙酯,40 ℃下吹干,加入2 mL乙腈,待凈化。
1.3.2 凈化
將1 mL上述待凈化液倒入萃取柱中,推拉柱塞桿將溶液過濾至2.5 mL離心管中,吸入萃取柱中,重復2次,加入1 mL乙腈淋洗柱中殘留的凈化液,二者合并在50 ℃下用氮氣至近干后,用甲醇-0.1%甲酸水(5/95,V/V)定容至1 mL,渦旋混勻30 s,用Nylon多層濾頭過濾后,用于LC-MS/MS檢測。
2 結果與討論
2.1 色譜條件的優化
硝基咪唑類抗生素是一類強極性化合物,在普通的反相色譜柱上很難被保留,而且峰形較差,影響定性和定量分析,選擇Ultimate NX-SCX強陽離子交換柱作為分析柱可大幅提高分析物保留性,而且各目標物均具有良好的峰形(圖1)。PFP五氟苯基柱可以滿足正相、反相和親水等分離的要求,尤其適合LCMS/MS,在分析苯并咪唑類抗生素方面具有一定的優勢,目標物的峰形和強度均優于傳統的C18反相色譜柱(圖2)。因此,選擇Ultimate NX-SCX柱分析硝基咪唑類抗生素,Ultimate NX-PFP柱分析苯并咪唑類抗生素。另外,比較了甲醇-水作和乙腈-水2個分離體系的效果,結果發現乙腈-水的分離效果較好。為改善峰形和靈敏度,在流動相中加入0.1%甲酸,靈敏度較高,峰形較好。因此,采用乙腈-0.1%甲酸水溶液作為流動相。
2.2 基質效應的評價
蜂蜜中16種硝基咪唑類和苯并咪唑類抗生素的提取和凈化主要采用乙酸乙酯提取后QuEChERS、m-PFC和MCX-SPE法等方法。如圖3所示,乙酸乙酯提取后未經任何凈化,該方法操作簡單,但基質干擾物多,適合大量樣品的篩查,不適合準確的定量分析,所產生的基質效應為30.4%~42.5%,存在強基質抑制現象。QuEChERS法是基于乙腈液-液提取、無水硫酸鎂和醋酸鈉鹽析分層,伯仲胺吸附劑PSA和反相吸附劑C18凈化后直接測定。該方法操作簡單,但凈化效果較差,樣液中仍然存在大量基質干擾物,16種生物堿基質效為49.0%~68.7%,依然存在明顯的基質抑制現象。m-PFC采用的多壁納米復合材料和MCX強陽離子柱凈化所產生的基質效應分別為80.4%~87.2%和79.6%~90.8%,基質抑制現象得到明顯改善。結果表明,多壁納米復合材料能有效降低基質效應,與MCX法效果相當,優于未凈化和QuEChERS法,對目標物具有良好的吸附作用。在進行SPE操作過程中,16種待測目標物與固相萃取填料之間發生分配或陽離子交換等作用時,必須嚴格控制樣液流速小于2 mL/min,才能使得目標物與吸附劑充分作用。因此,SPE法普遍存在操作時間長(活化、上樣、淋洗和洗脫)和操作難度較大(復雜基質堵塞篩板)等問題,明顯降低實際工作效率。與SPE法相比,m-PFC可通過反復抽拉的凈化方式除去樣液中的雜質,保證吸附劑與雜質之間的充分作用,目標物依然保留在提取液中,同時降低操作難度,提高工作效率。
2.3 工作曲線和相關系數
為彌補基質的基質抑制效應,試驗采用基質空白工作曲線法進行定量分析。由表2可知,16種硝基咪唑類和苯并咪唑類抗生素在各自線性范圍內具有良好的線性關系,線性相關系數r≥0.995 0;方法檢出限(S/N=3)為0.03~0.5 μg/kg,定量限(S/N=10)為0.1~1.0 μg/kg。
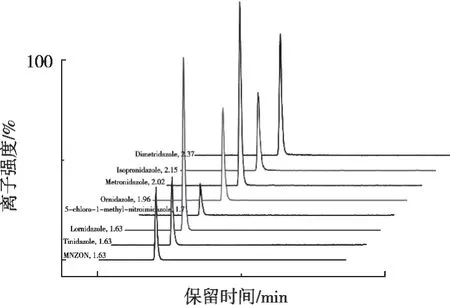
圖1 8種硝基咪唑類抗生素總離子流圖(TIC)

圖2 8種苯并咪唑類抗生素總離子流圖(TIC)
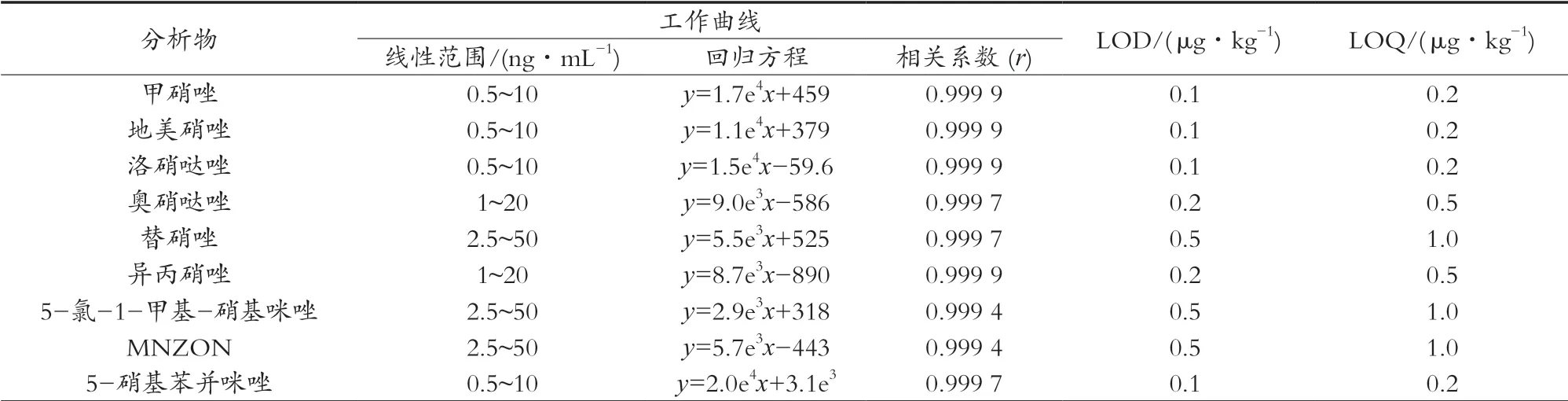
表2 16種硝基咪唑類和苯并咪唑類抗生素的工作曲線、檢出限和定量限
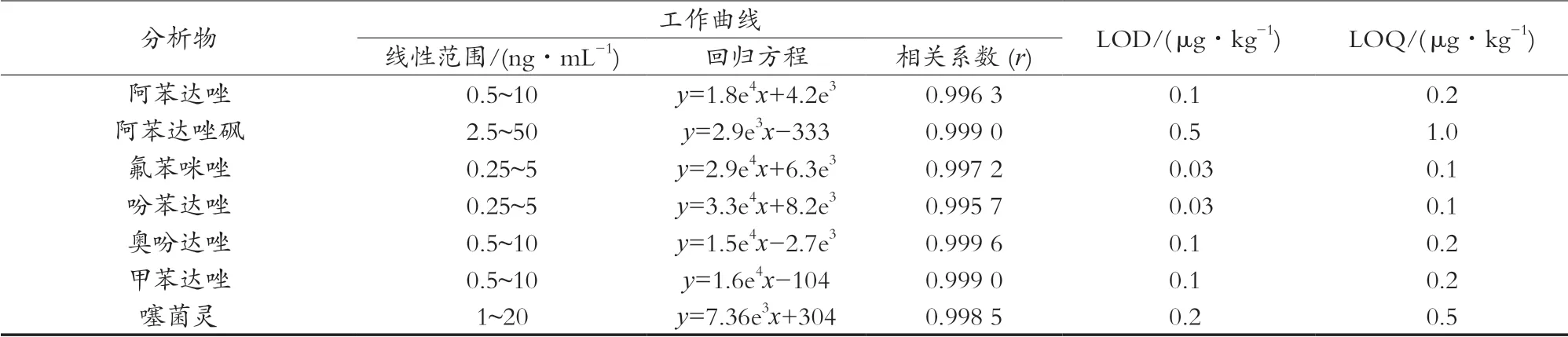
接表2
2.4 方法的回收率及精密度
分別取基質空白樣品,進行加標試驗,設3個加標水平:1倍LOQ、2倍LOQ和10倍LOQ,上機測定。結果表明,以空白蜂蜜作為加標基質的回收率為80.0%~106.8%,相對偏差(δRSD,n=6)分別為4.1%~6.7%,滿足蜂蜜中硝基咪唑類和苯并咪唑類抗生素殘留檢測的要求。
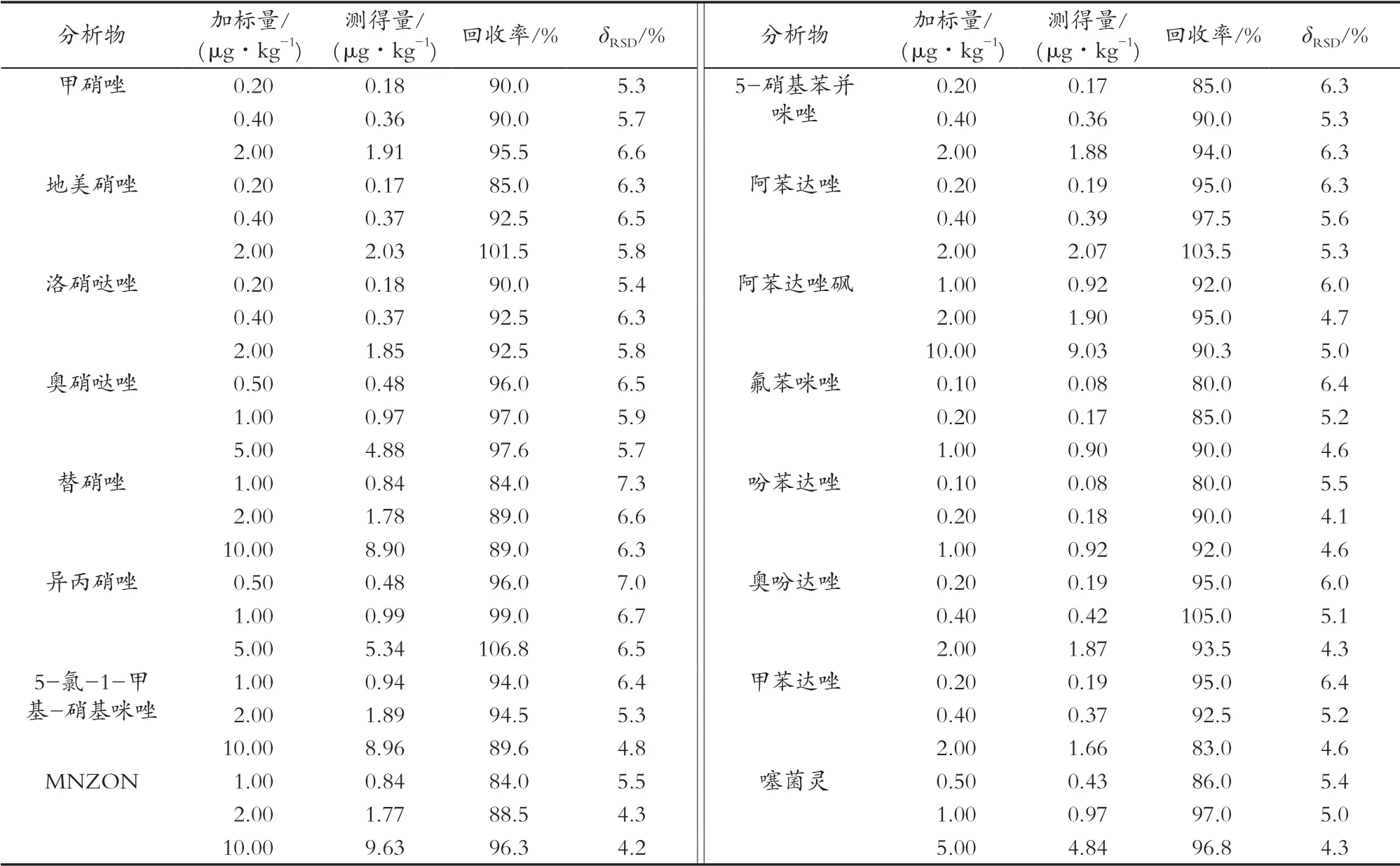
表3 蜂蜜中16種硝基咪唑類和苯并咪唑類抗生素的加標回收率和相對標準偏差
2.5 實際樣品的檢測
用試驗方法對2018和2019年寧夏省風險監測項目抽檢的30份蜂蜜進行檢測,其中2份樣本(2/30)中檢出甲硝唑,含量為1.22和1.78 μg/kg;其余蜂蜜樣本(28/30)中硝基咪唑類和苯并咪唑類抗生素的含量均低于方法檢出限(0.03~0.5 μg/kg)。
3 結論
采用m-PFC凈化,分別選用強陽離子交換柱和五氟苯基柱分離分析,LC-MS/MS測定蜂蜜中16種硝基咪唑類和苯并咪唑類抗生素的含量,回收率(80.0%~106.8%)和精密度(<7.0%)符合相關檢測要求。試驗方法快速簡便、靈敏度高、重現性好,對于加強寧夏地區該項目的監測工作具有重要意義。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
