蒙藥哈日-沙布嘎(鐵桿蒿)的質量標準提升
2021-04-07 04:15:20袁小紅龐克堅唐輝張虹魏灃劉樂樂姜國珍
中國藥房 2021年5期
袁小紅 龐克堅 唐輝 張虹 魏灃 劉樂樂 姜國珍

摘 要 目的:提升蒙藥哈日-沙布嘎(鐵桿蒿)的質量標準,為全面評價其質量提供科學依據。方法:對哈日-沙布嘎進行外觀性狀鑒別和顯微鑒別;采用薄層色譜(TLC)法對藥材中的東莨菪苷、綠原酸、咖啡酸、東莨菪內酯和3,5-二咖啡酰奎寧酸進行定性鑒別;建立高效液相色譜(HPLC)法測定藥材中上述5種成分的含量;并對藥材的水分、總灰分和浸出物進行檢查。結果:哈日-沙布嘎藥材莖呈類圓柱形,表面呈紫色或紫褐色或青褐色;葉呈卵形或矩圓狀卵形,氣香;花黃色,頭狀花序,近球形或半球形;粉末呈綠色或黃綠色,花粉粒具3個萌發孔,薄壁細胞簇晶棱尖銳,螺紋導管眾多,偶見具緣紋孔導管,木纖維多成束存在。TLC結果顯示,供試品在與5種對照品色譜相應位置上均顯相同顏色的斑點。東莨菪苷、綠原酸、咖啡酸、東莨菪內酯、3,5-二咖啡酰奎寧酸的質量濃度分別在85.60~428.00、10.16~101.60、10.20~102.00、40.84~408.40、40.80~408.00 μg/mL范圍內與各自峰面積呈良好線性關系(r均大于0.999 0);精密度、穩定性、重復性試驗的RSD均小于3.00%(n=6);平均加樣回收率分別為103.07%、99.66%、98.37%、97.78%、98.40%(RSD均小于3.00%,n=6)。10批藥材樣品中上述5種成分含量范圍分別為0.36%~1.23%、0.09%~0.51%、0.04%~0.13%、0.61%~1.13%、0.12%~1.11%;水分、總灰分、水溶性浸出物的平均含量分別為6.25%、5.86%、26.50%。結論:該研究在哈日-沙布嘎原質量標準的基礎上增加了顯微鑒別、TLC鑒別、含量測定以及水分、總灰分和浸出物等檢查項,方法精密、準確、穩定性良好,能夠為更加科學、規范地評價該藥材質量提供依據。
關鍵詞 蒙藥;哈日-沙布嘎(鐵桿蒿);質量標準;顯微鑒別;薄層色譜法;高效液相色譜法;東莨菪苷;綠原酸;咖啡酸;東莨菪內酯;3,5-二咖啡酰奎寧酸
中圖分類號 R284.1 文獻標志碼 A 文章編號 1001-0408(2021)05-0536-06
ABSTRACT OBJECTIVE: To improve the quality standard of Mongolian medicine Artemisia sacrorum, and to provide scientific basis for comprehensive quality evaluation. METHODS: The appearance and microscopic characteristics of A. sacrorum were identified; scopoletin, chlorogenic acid, caffeic acid, scopoletin and 3,5-dicaffeoylquinic acid were identified quantitatively by TLC; the contents of above 5 components were determined by HPLC. The water content, total ash and extract were examined. RESULTS: The stem of A. sacrorum was cylindrical, and its surface was purple or purple-brown or cyan-brown; the leaves were ovate or oblong-ovate, fragrant; the flowers were yellow, head-shaped, subglobose or hemispherical. The powder was green or yellow-green,its pollen grain had three germination; the parenchymal cell clusters with sharp edges and numerous threaded ducts,occasionally having marginal pitted ducts; its wood fibers were in bundles mostly. Results of TLC showed that the spots of the same color were found in the corresponding positions of chromatogram for 5 substance control and samples. The linear range of scopoletin, chlorogenic acid, caffeic acid, scopolactone and 3,5-dicaffeoylquinic acid were 85.60-428.00, 10.16-101.60, 10.20-102.00, 40.84-408.40 and 40.80-408.00 μg/mL (all r>0.999 0). RSDs of precision, stability, repeatability tests were all less than 3.00% (n=6). The average recoveries were 103.07%,99.66%,98.37%,97.78%, 98.40% (all RSDs<3.00%, n=6). The contents of the above-mentioned 5 compounds in 10 batches of samples were 0.36%-1.23%, 0.09%-0.51%, 0.04%-0.13%, 0.61%-1.13%, 0.12%-1.11%, respectively; the average contents of water, total ash and water soluble extract were 6.25%, 5.86%, 26.50%, respectively.? CONCLUSIONS: On the basis of the original quality standard of A. sacrorum, microscopic identification, TLC identification, content determination and examination items of water, total ash and extract are added. The method shows good precision, accuracy and stability, which can provide reference for more scientific and standardized evaluation of the quality of this medicinal material.
KEYWORDS Mongolian medicine; Artemisia sacrorum; Quality standard; Microscopic identification; TLC; HPLC; Scopoletin; Chlorogenic acid; Caffeic acid; Scopoletin; 3,5-dicaffeoylquinic acid
蒙藥哈日-沙布嘎,為菊科植物鐵桿蒿Artemisia sacrorum Ledeb.的干燥全草,別名白蓮蒿、萬年蒿;其在新疆、寧夏、甘肅、西藏、內蒙古等地均有廣泛分布[1],且用藥多為自采。哈日-沙布嘎全株均能入藥,據蒙醫藥有關書籍記載,其性涼、味苦,具有殺蟲止痛、消腫、燥“協日烏素”(注:“協日烏素”為蒙醫對病癥“黃水”的專有名詞)等功能[2]。研究發現,哈日-沙布嘎的主要成分有黃酮類、肉桂酸類、香豆素類、萜類、揮發油等[3-5]。其提取物中含有的木脂素具有較好的保肝作用;香豆素類化合物具有抗炎、抗菌等作用;地上部分的甲醇提取物顯示出強抗氧化作用[6-8];此外,其含有的咖啡酰奎尼酸類化合物具有較強的抗氧化、抗炎活性[9],這一作用與哈日-沙布嘎用藥功效一致。
哈日-沙布嘎收載于1998年版的《衛生部藥品標準·蒙藥分冊》中,但該標準僅收錄了該藥材的外觀性狀、性味和功能主治等,缺乏其他鑒別項,也沒有基于藥效的化學成分含量測定[2]。現有對其質量控制的報道多是采用高效液相色譜(HPLC)法對藥材中單一成分進行定量分析,如徐犇等[10]對藥材中東莨菪內酯進行了含量測定,佟皎薇等[11]測定了藥材中綠原酸的含量,這些方法均難以全面評價藥材質量。鑒于此,本研究采集了10批哈日-沙布嘎藥材,在原質量標準基礎上增加了顯微鑒別、薄層色譜(TLC)鑒別、含量測定以及水分、總灰分和浸出物等檢查項,以期對該藥材的質量進行全面控制。
1 材料
1.1 主要儀器
本研究所使用的主要儀器有:Primo Star型數碼生物顯微鏡(德國Zeiss公司)、Eclipse E200型生物顯微鏡(日本Nikon公司)、YOKO-ZS型薄層數碼成像儀(武漢藥科新技術開發有限公司)、G型硅膠板(青島海洋化工有限公司,規格50 mm×100 mm,批號20191109)、展開槽(北京杰瑞恒達科技有限公司,規格110 mm×120 mm)、e2695型HPLC儀(含2998 型光電二極管矩陣檢測器,美國Waters公司)、KH-300DE型臺式數控超聲波清洗器(昆山禾創超聲儀器有限公司)、BT125D型十萬分之一電子分析天平(德國Sartorius公司)、UV-2600型紫外分光光度計(日本Shimadzu公司)。
1.2 主要試劑
本研究所使用的主要試劑有:東莨菪苷對照品、綠原酸對照品、咖啡酸對照品、東莨菪內酯對照品、3,5-二咖啡酰奎寧酸對照品(上海源葉生物科技有限公司,批號分別為Z15J11S115885、Y24J7K16726、Y09J8C28349、Z17J10Y90380、Z05M10X87215,純度均大于98%),乙腈(美國Fisher公司,色譜純),磷酸、甲醇(天津市富宇精細化工有限公司,分析純),超純水。
1.3 藥材
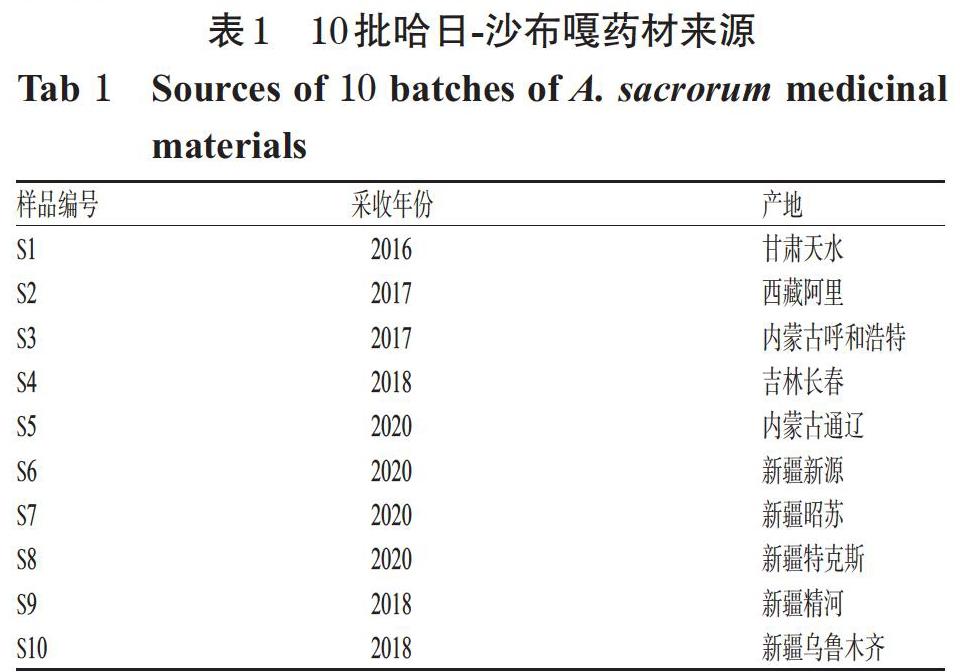
10批哈日-沙布嘎藥材來源見表1。其中S1~S2號藥材采購于新疆精河縣蒙醫診所, S3~S5號藥材采購于新疆博爾塔拉蒙古自治州蒙醫醫院,S6~S10號藥材采自新疆新源、昭蘇、烏魯木齊等地,均經新疆博爾塔拉蒙古自治州蒙醫醫院巴特孟克主任醫師鑒定為菊科植物鐵桿蒿A. sacrorum Ledeb.的干燥全草。
2 方法與結果
2.1 溶液的制備
2.1.1 對照品溶液的制備 分別取東莨菪苷、綠原酸、咖啡酸、東莨菪內酯、3,5-二咖啡酰奎寧酸對照品適量,精密稱定,加入甲醇制成上述5個成分質量濃度分別為1.070、1.016、1.020、2.042、2.040 mg/mL的單一成分對照品貯備液。分別取東莨菪苷對照品貯備液0.75 mL,綠原酸、咖啡酸對照品貯備液各0.20 mL,東莨菪內酯、? ? 3,5-二咖啡酰奎寧酸對照品貯備液各0.40 mL,置于同一5 mL量瓶中,用甲醇稀釋至刻度,混勻,制成混合對照品溶液,備用。
2.1.2 供試品溶液的制備 取哈日-沙布嘎藥材粉末(過30目篩)0.5 g,精密稱定,置于100 mL錐形瓶中,加甲醇20 mL,稱質量,超聲(功率300 W,頻率40 kHz)處理1 h,取出,放冷至室溫,再次稱質量,用甲醇補足減失的質量,先后以濾紙、0.45 μm微孔濾膜濾過,取續濾液,即得供試品溶液。
2.1.3 陰性對照溶液的制備 取甲醇20 mL,置于100 mL錐形瓶中,按“2.1.2”項下方法制成陰性對照溶液。
2.2 性狀和顯微鑒別
2.2.1 性狀鑒別 藥材莖呈類圓柱形,直徑1~5 mm,表面呈紫色或紫褐色或青褐色;質韌易折斷,斷面呈綠白色或黃褐色,部分莖中空。葉呈卵形或矩圓狀卵形,二回羽狀全裂;側裂片2~5對,羽狀全裂;小裂片條狀披針形,寬1~3 mm,全緣;上表面呈綠色,下表面呈灰綠色,氣香。花黃色,頭狀花序,近球形或半球形,直徑2~3 mm,下垂,排列成復總狀花序。哈日-沙布嘎藥材的外觀性狀見圖1。
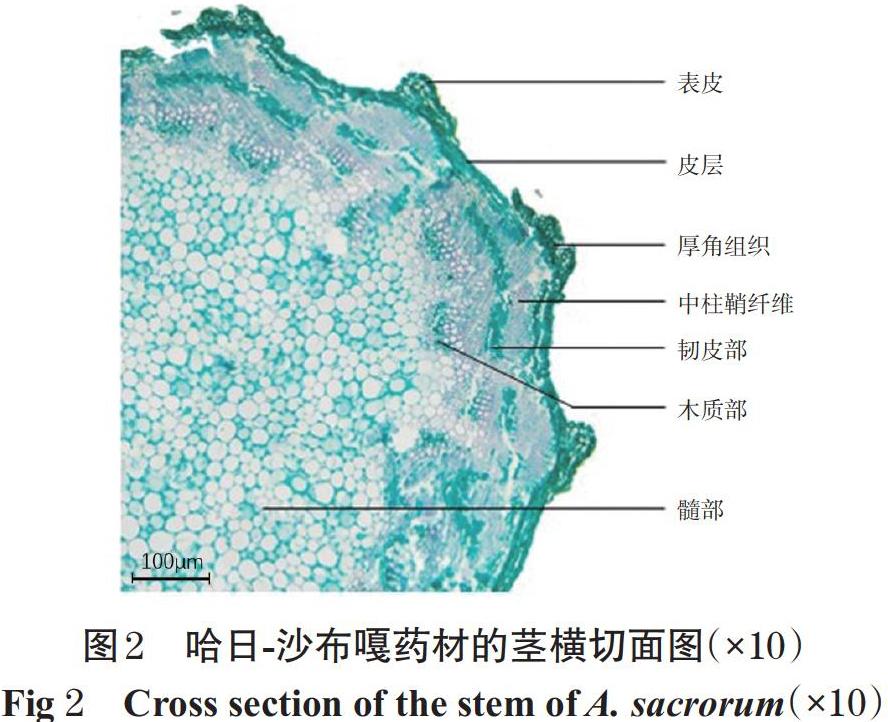
2.2.2 莖橫切面顯微鑒別 藥材的莖橫切面有1列表皮細胞,呈類方形或圓形;皮層由多層薄壁細胞組成,呈類橢圓形或不規則形,在縱棱處有1~3層厚角組織;維管束為外韌型,由1~4列纖維構成帽狀纖維束;韌皮部薄壁細胞呈圓形或橢圓形,形成層不明顯,導管排列不規則;髓部較寬廣,由圓形或橢圓形的薄壁細胞組成。哈日-沙布嘎藥材的莖橫切面圖見圖2。
2.2.3 粉末顯微鑒別 藥材粉末呈綠色或黃綠色。粉末中可見大量花粉粒,具3個萌發孔,散在,直徑16.5~21.1 μm;草酸鈣簇晶聚集于薄壁細胞內,棱尖銳,直徑70.8~117.0 μm;非腺毛多單細胞,細長,末端尖銳;螺紋導管眾多,偶見具緣紋孔導管;木纖維多成束存在,壁不甚厚、斷裂、破碎;莖表皮細胞呈綠色,類圓形,排列整齊;葉表皮細胞的細胞壁呈微波狀,細長。哈日-沙布嘎粉末特征圖見圖3。
2.3 TLC鑒別
按照2020年版《中國藥典》(四部)TLC法(通則0502)實驗操作要求[12],分別吸取“2.1”項下各單一成分對照品貯備液5 μL和供試品溶液10 μL,點于同一硅膠G薄層板上,在常溫下飽和后展開,取出,晾干,檢視。其中,東莨菪苷的展開劑為三氯甲烷-甲醇(7 ∶ 3,V/V),置于紫外燈365 nm波長下檢視;綠原酸的展開劑為乙酸乙酯-丙酮-甲酸-水(8 ∶ 3 ∶ 1 ∶ 1,V/V/V/V),噴以三氯化鐵乙醇溶液顯色后加熱,在日光燈下檢視;咖啡酸的展開劑為甲苯-乙酸乙酯-甲酸(7 ∶ 3 ∶ 0.5,V/V/V),噴以三氯化鋁乙醇溶液顯色后置于紫外燈365 nm波長下檢視;東莨菪內酯的展開劑為乙酸乙酯-甲醇(15 ∶ 1,V/V),置于紫外燈365 nm波長下檢視;3,5-二咖啡酰奎寧酸的展開劑為乙酸乙酯-丙酮-甲酸-水(8 ∶ 3 ∶ 1 ∶ 0.4,V/V/V/V),噴以三氯化鋁乙醇溶液顯色后置于紫外燈365 nm波長下檢視。結果,供試品與各對照品在相應的位置上,顯相同顏色的斑點,詳見圖4。
2.4 含量測定
2.4.1 色譜條件 以ACE C18 PFP(250 mm×4.6 mm,5? μm)為本試驗色譜柱,以乙腈(A)-0.1%磷酸溶液(B)為流動相,梯度洗脫(0~5 min,13%A→15%A;5~8 min,15%A→18%A;8~20 min,18%A→25%A;20~25 min,25%A→55%A),流速為1 mL/min,檢測波長為328 nm,柱溫為30 ℃,進樣量為5 μL。
2.4.2 系統適用性試驗 取“2.1”項下混合對照品溶液、供試品溶液、陰性對照溶液各5 μL,按“2.4.1”項下色譜條件進樣測定,記錄色譜圖(見圖5)。結果表明,在該色譜條件下,理論板數按東莨菪內酯峰計算不低于20 000,各色譜峰與相鄰峰的分離度均大于1.2,陰性對照對各成分的測定無干擾。
2.4.3 線性關系考察 分別取東莨菪苷對照品貯備液0.40、0.50、0.75、1.00、1.50、2.00 mL,綠原酸、咖啡酸對照品貯備液各0.05、0.10、0.20、0.30、0.40、0.50 mL,東莨菪內酯、3,5-二咖啡酰奎寧酸對照品貯備液各0.10、0.20、0.40、0.60、0.80、1.00 mL,分別置于6個5 mL量瓶中,用甲醇稀釋至刻度,混勻,配制成系列濃度的混合對照品溶液。按“2.4.1”項下色譜條件分別進樣測定,記錄峰面積。以質量濃度(x)為橫坐標、峰面積(y)為縱坐標,繪制標準曲線,計算得5種成分的線性回歸方程、線性范圍、相關系數,結果見表2。
2.4.4 檢出限與定量限考察 取“2.1.1”項下各對照品貯備液適量,用甲醇逐步稀釋,按“2.4.1”項下色譜條件進樣測定,記錄峰面積,分別以信噪比3 ∶ 1、10 ∶ 1測定檢出限、定量限,結果見表2。
2.4.5 精密度試驗 精密吸取“2.1.1”項下混合對照品溶液,按“2.4.1”項下色譜條件連續進樣測定6次,記錄5個成分的峰面積。結果,東莨菪苷、綠原酸、咖啡酸、東莨菪內酯、3,5-二咖啡酰奎寧酸峰面積的RSD分別為0.59%、0.43%、0.38%、0.27%、0.35%(n=6),表明儀器精密度良好。
2.4.6 重復性試驗 分別稱取同一批樣品(編號S3)共6份,每份0.5 g,精密稱定,置于100 mL錐形瓶中,按“2.1.2”項下方法制備供試品溶液,再按“2.4.1”項下色譜條件進樣測定,記錄5個成分的峰面積,代入標準曲線計算樣品含量。結果,樣品中東莨菪苷、綠原酸、咖啡酸、東莨菪內酯、3,5-二咖啡酰奎寧酸的平均含量分別為0.54%、0.15%、0.08%、0.61%、0.53%,RSD分別為2.05%、2.74%、2.23%、2.44%、2.80%(n=6),表明方法重復性良好。
2.4.7 穩定性試驗 取同一份供試品溶液(編號S1),分別于室溫下放置0、2、4、8、12、24 h時按“2.4.1”項下色譜條件進樣測定,記錄5個成分的峰面積。結果,東莨菪苷、綠原酸、咖啡酸、東莨菪內酯、3,5-二咖啡酰奎寧酸峰面積的RSD分別為2.32%、2.67%、1.19%、1.86%、1.48%(n=6),表明供試品溶液在室溫下放置24 h內穩定性良好。
2.4.8 加樣回收率試驗 稱取已知含量的樣品(編號S4)共6份,每份0.5 g,精密稱定,精密加入“2.4.3”項下最后一個系列濃度的混合對照品溶液適量,按“2.1.2”項下方法制備供試品溶液,再按“2.4.1”項下色譜條件進樣測定,記錄峰面積,計算加樣回收率,結果見表3。由表3可知,東莨菪苷、綠原酸、咖啡酸、東莨菪內酯、3,5-二咖啡酰奎寧酸加樣回收率的RSD分別為1.24%、1.32%、2.04%、2.64%、1.95%(n=6),表明方法準確度良好。
2.4.9 樣品含量測定 取10批藥材樣品各適量,分別按“2.1.2”項下方法制備供試品溶液,再按“2.4.1”項下色譜條件進樣測定,記錄峰面積,代入標準曲線計算5種成分含量。每批樣品平行測定3次,取平均值,結果見表4。
2.5 檢查
2.5.1 水分、總灰分測定 分別按照2020年版《中國藥典》(四部)通則0832(烘干法)和通則2302進行水分和總灰分測定[12]。每批樣品平行測定3次,取平均值。結果,各批樣品水分含量為5.85%~6.58%,平均值為6.25%;各批樣品總灰分含量為4.87%~6.57%,平均值為5.86%,詳見表5。以平均值上浮約20%,暫定藥材水分含量不得超過7.50%、總灰分含量不得超過7.03%。
2.5.2 浸出物測定 按照2020年版《中國藥典》(四部)通則2201[12],采用水為溶劑以熱浸法提取藥材,測定水溶性浸出物的含量。每批樣品平行測定3次,取平均值。結果,各批樣品水溶性浸出物含量為21.26%~33.53%,平均值為26.50%,詳見表5。以平均值下浮約20%,暫定藥材水溶性浸出物含量不得低于21.20%。
3 討論
3.1 樣品提取條件的選擇
在樣品的提取中,筆者對提取溶劑(甲醇、50%甲醇、乙醇、50%乙醇)、料液比(1 ∶ 40、1 ∶ 20、1 ∶ 10、1 ∶ 5,? ? g/mL)、提取時間(0.5、1、2、3 h)進行了篩選,綜合考慮所得色譜峰的峰面積、分離度、峰形及制備方法的簡便、快捷,最終確定本研究提取條件為提取溶劑甲醇、料液比 1 ∶ 40(g/mL)、提取時間1 h。
3.2 TLC法展開劑的選擇
筆者在預試驗中,選用不同展開系統對不同批次藥材進行了TLC鑒別。綠原酸的展開劑考察了乙酸乙酯-甲酸-水(7 ∶ 2.5 ∶ 2.5,V/V/V)、乙酸乙酯-丁酮-甲酸-水? (10 ∶ 6 ∶ 1 ∶ 2,V/V/V/V)、乙酸乙酯-丙酮-甲酸-水(8 ∶ 3 ∶ 1 ∶ 1,V/V/V/V);咖啡酸的展開劑考察了甲苯-乙酸乙酯-甲酸(7 ∶ 3 ∶ 0.5,V/V/V)、甲苯-乙酸乙酯-甲酸(5 ∶ 3 ∶ 1,V/V/V)、甲苯-乙酸乙酯-甲酸(15 ∶ 3.5 ∶ 0.5,V/V/V);東莨菪苷的展開劑考察了三氯甲烷-甲醇(6 ∶ 4,V/V)、三氯甲烷-甲醇(8 ∶ 2,V/V)、三氯甲烷-甲醇(7 ∶ 3,V/V);東莨菪內酯的展開劑考察了乙酸乙酯-甲醇(15 ∶ 1,V/V)、乙酸乙酯-甲醇-濃氨試液(17 ∶ 2 ∶ 1,V/V/V);3,5-二咖啡酰奎寧酸的展開劑考察了乙酸乙酯-丙酮-甲酸-水(7 ∶ 3 ∶ 1 ∶ 1.2,V/V/V/V)、乙酸乙酯-甲酸-水(7 ∶ 2.5 ∶ 2.5,V/V/V)、乙酸乙酯-丙酮-甲酸-水(8 ∶ 3 ∶ 1 ∶ 0.4,V/V/V/V)。結果顯示,采用文中展開劑能較好地將主斑點與其他斑點分離。
3.3 HPLC法檢測波長的選擇
結合HPLC圖和預試驗結果,可知5種成分中東莨菪內酯含量較高,當東莨菪內酯有最大吸收時,其他成分均有較強吸收,且變換檢測波長對東莨菪內酯影響較大,對其他成分影響相對較小,故選用東莨菪內酯來篩選檢測波長。
取東莨菪內酯對照品貯備液適量,以甲醇稀釋成質量濃度為0.016 mg/mL的溶液,用紫外可見分光光度計在200~600 nm波長范圍內掃描,結果可見其在334 nm波長處有最大吸收。再結合文獻中東莨菪內酯的檢測波長[10,13],比較供試品溶液在328、334 nm波長下的色譜圖數據,結果表明在328 nm波長下,5種成分的色譜峰基線均平穩,且均有較強吸收,故選擇328 nm為檢測波長。
3.4 HPLC法流動相的選擇
在選擇HPLC法流動相時,筆者以各色譜峰分離度、出峰時間、峰形等為評價指標,比較了甲醇-0.1%甲酸溶液、甲醇-0.1%磷酸溶液、乙腈-0.1%磷酸溶液、乙腈-0.1%甲酸溶液4種流動相系統。結果表明,以甲醇-水溶液系統為流動相梯度洗脫時,柱壓較高、峰形較差;以乙腈-0.1%甲酸溶液梯度洗脫時,東莨菪苷與綠原酸分離度較差;以乙腈-0.1%磷酸溶液梯度洗脫時,柱壓較低,各色譜峰分離良好且基線平穩,故選定乙腈-0.1%磷酸溶液作為梯度洗脫流動相。
綜上所述,本研究在哈日-沙布嘎原質量標準的基礎上增加了顯微鑒別、TLC鑒別、含量測定以及水分、總灰分和浸出物等檢查項,首次對哈日-沙布嘎中東莨菪苷、咖啡酸、3,5-二咖啡酰奎寧酸進行了含量測定,方法精密、準確、穩定性良好,能夠為更加科學、規范地評價蒙藥哈日-沙布嘎的質量提供依據。
參考文獻
[ 1 ] 曾育麟,李星煒.中國民族藥志:第4卷[M].成都:四川民族出版社,2007:514.
[ 2 ] 衛生部藥典委員會.中華人民共和國衛生部藥品標準:蒙藥分冊[S].北京:人民衛生出版社,1998:38.
[ 3 ] 王青虎,吳榮君,齊格奇,等.蒙藥鐵桿蒿化學成分研究[J].中國藥學雜志,2015,50(16):1380-1383.
[ 4 ] 劉丹.萬年蒿花序化學成分的研究[D].吉林:延邊大學,2015.
[ 5 ] 玉華,王青虎,韓那仁朝克圖,等.蒙藥鐵桿蒿石油醚提取物中6個萜類化合物的分離與鑒定[J].中國藥物化學雜志,2016,26(2):142-145.
[ 6 ] 孫艷濤,蔣黃卉,趙磊,等.朝鮮族藥材萬年蒿地上部分化學成分研究[J].中國藥學雜志,2020,55(10):806-810.
[ 7 ] 張明曉,黃國英,白羽琦,等.南、北五味子的化學成分及其保肝作用的研究進展[J/OL].中國中藥雜志,2020[2021- 02-01].https://doi.org/10.19540/j.cnki.cjcmm.20201127. 601. DOI:org/10.19540/j.cnki.cjcmm.20201127.601.
[ 8 ] 王陳萍,許波,蔡向明,等.不同五味子類木脂素成分對損傷肝細胞的保護作用比較[J].中國醫藥導報,2017,14(19):35-38.
[ 9 ] 吳琪珍.咖啡酰奎寧酸類化合物研究進展[J].中國野生植物資源,2020,39(4):48-53、60.
[10] 徐犇,李納,劉吉爽,等.吉林省不同產地萬年蒿中東莨菪內酯HPLC含量測定[J].長春中醫藥大學學報,2018,34(5):880-883.
[11] 佟皎薇,李佳林. HPLC法測定蒙藥材鐵桿蒿不同部位中綠原酸的含量[J].中國民族醫藥雜志,2015,21(6):39.
[12] 國家藥典委員會.中華人民共和國藥典:四部[S]. 2020年版.北京:中國醫藥科技出版社,2020:59、114、232、234.
[13] 國家藥典委員會.中華人民共和國藥典:一部[S]. 2020年版.北京:中國醫藥科技出版社,2020:147.
(收稿日期:2020-10-15 修回日期:2021-02-08)
(編輯:胡曉霖)
