三氟甲苯丁二酮合成方法研究進展
2021-04-10 01:56:50張春光李培杰陳永龍
天津化工 2021年2期
張春光,李培杰,陳永龍
(鄭州瑞普生物工程有限公司,河南 鄭州450007)
三氟甲苯丁二酮(C11H9F3O2),又稱4,4,4-三氟-1-(4-甲苯基)-1,3-丁二酮,白色晶體,不溶于水,易溶于乙醇、甲醇、丙酮等有機溶劑。一種比較重要的醫藥中間體,主要用于抗關節炎藥塞來昔布的合成。目前常見的合成方法仍然是以三氟乙酸乙酯為原料與對甲基苯乙酮經過縮合反應再經酸化制得三氟甲苯丁二酮。因此,本文主要從三氟乙酸乙酯、三氟乙酸甲酯及其他原料與對甲基苯乙酮在不同縮合劑作用下通過Claisen反應制備三氟甲苯丁二酮的合成方法,并對其反應機理進行分析,得出了不同反應方法的優缺點,提出了一些個人觀點。
1 由三氟乙酸乙酯制三氟甲苯丁二酮
最早合成三氟甲苯丁二酮的Penning[1]在1994年以甲醇作為溶劑,氬氣保護,對甲基苯乙酮與三氟乙酸乙酯在25%甲醇鈉-甲醇溶液的作用下,以投料比n(酮)∶n(酯)∶n(鈉)=1∶1.18∶1.34投料通過Claisen縮合得到,采用鹽酸中和,得到棕色油狀物,收率為94%。且該還報道了三氟氯苯丁二酮的有關合成,其中采用甲基叔丁基醚作為溶劑,反應16 h,得到產物,收率85%。這兩個反應均屬于Claisen縮合反應。反應式如圖1所示:
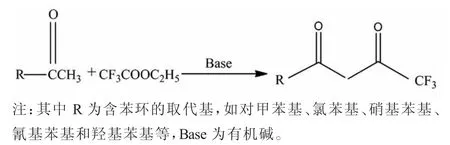
圖1 Claisen縮合反應反應式
2000年梅之南[2]在Penning的基礎上進行了改進:用甲苯代替甲醇為溶劑,用自制乙醇鈉代替甲醇鈉為縮合劑,在100℃反應4h。脫甲苯,然后加冰乙酸及冰的混合液來淬滅反應,乙酸乙酯萃取4次,無水硫酸鈉干燥,蒸出溶劑直接得到棕色固體,不需要再純化,而且產度較高,收率高達94.3%。2001年王恩舉[3]在Penning的基礎上首先改變了加料順序,混合甲醇鈉溶液與對甲基苯乙酮時,將加料方式改為將對甲基苯乙酮滴加到甲醇鈉溶液中,其反應狀態由回流改為45℃密閉反應24h。同時也降低了三氟乙酸乙酯的揮發,收率提高為96%。該方法主要優點就是反應條件較溫和,降低了耗能,提高了收率。2002年張邦樂[4]在Penning基礎上采用非質子溶劑乙醚為溶劑,縮短了反應時間,同時對加料方式和順序進行了改變,先將甲醇鈉與三氟乙酸乙酯混合攪拌,再滴加對甲基苯乙酮,投料比n(酮)∶n(酯)∶n(醇鈉)=1∶1∶1.1,回流10h,收率高達99.1%。改變加料順序使對甲基苯乙酮與醇鈉的接觸時間短,抑制了酮的自身縮合,降低了不良反應。2003年梅世昌[5]采用乙醇鈉作為縮合劑,甲苯作為溶劑,對甲基苯乙酮和三氟乙酸乙酯為原料,100℃反應5h,制得三氟甲苯丁二酮,收率為89.1%。另外該專利還報道了對甲氧基苯乙酮、對甲硫基苯乙酮、對甲氨基苯乙酮和二甲氨基苯乙酮與三氟乙酸乙酯的縮合反應。其基本原理見圖2:

圖2 縮合反應原理圖
2005年Li M H[6]在Penning基礎上用乙醇鈉代替甲醇鈉作為縮合劑,乙醇作為溶劑,氮氣保護,且反應時間從24h縮短至4h,得到89%的收率。2007年Marie[7]采用四氫呋喃為溶劑,氫化鈉作為縮合劑,在-5~0℃向溶解有對甲基苯乙酮的四氫呋喃溶液中滴加氫化鈉,然后再滴加三氟乙酸乙酯,然后在室溫下反應5h,反應結束冰鹽酸淬滅有機物調節pH,乙酸乙酯萃取,干燥,濃縮,固體殘余物用正己烷沖洗,減壓干燥得到團狀固體,然后溶于二氯甲烷中,打漿,干燥得到三氟甲苯丁二酮,收率為95%。該方法反應條件溫和,耗能較低,收率較高,產物純度高,不足之處就是氫化鈉價格比較昂貴。2008年Gyorgy[8]參照Penning的方法進行了改進,以甲醇鈉作為縮合劑,對甲基苯乙酮和三氟乙酸乙酯為原料,以n(酮)∶n(酯)∶n(鈉)=1∶1.04∶1.67的投料比在80℃反應10h,得到的鈉鹽用鹽酸來酸化處理,最后得到產物,收率為95%。2010年Ambati[9]在采用甲苯作為溶劑,甲醇鈉為縮合劑,對甲基苯乙酮和三氟乙酸乙酯為原料,以n(酮)∶n(酯)∶n(鈉)=1∶1.2∶1.2的投料比在55~60℃反應4h,鹽酸調節pH,最終得到93.3%的收率。
2011年儲剛[10]在專利CN102863386A中將對甲基苯乙酮以及作為溶劑的甲醇混合,在有機堿作用下,滴加三氟乙酸乙酯,60~80℃下反應11~14h,濃縮脫溶,鹽酸調pH 2~4,萃取,有機相濃縮得到產物。該專利采用三氟乙酸乙酯大過量的方式來促進對甲基苯乙酮反應徹底,提高產率,縮短了反應時間。蔣惠綱[11]采用對甲基苯乙酮和三氟乙酸乙酯在堿作用下在非質子溶劑中進行縮合得到三氟甲苯丁二酮,分別采用了氫化鈉、氫化鉀作為縮合劑進行了反應,甲苯為溶劑,其中在氫化鈉作用下,65℃下反應1h,得到96%的收率。該方法優點在于反應條件溫和,反應時間短,操作簡單,產物純度高。2013年陳錦華[12]以無水乙醇代替甲醇為溶劑,氬氣保護下,分別選用甲醇鈉、乙醇鈉和氫化鈉作為縮合劑,進行縮合反應,60℃回流反應24h,鹽酸調節pH,乙酸乙酯萃取分離,然后經過旋蒸,干燥,得到紅棕色油狀液產物,結果見表1,實驗結果表明當以投料比n(酮)n(鈉)=1∶1.6進行反應,選用甲醇鈉作為縮合劑時,對甲基苯乙酮的轉化率及選擇性較好,產物收率較高,收率大概為74.3%。
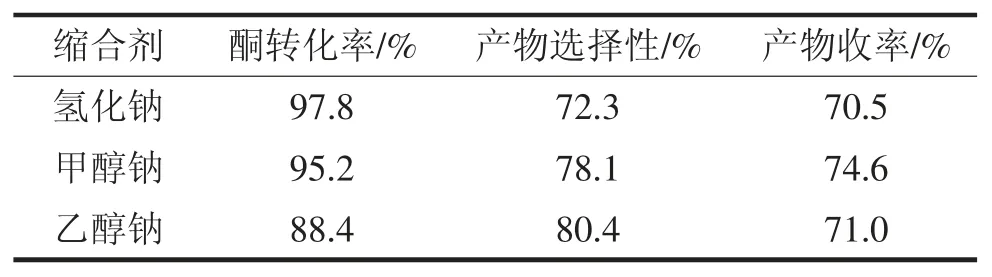
表1縮合劑對轉化率、選擇性、收率的影響
苗青[13]在專利CN 103102306 A中采用對甲基苯乙酮與三氟乙酸乙酯以甲醇鈉為催化劑,以甲基叔丁基醚作為溶劑,升溫20~100℃回流反應12h,加鹽酸,得到產物,收率為99.1%。大大縮短了反應時間,且收率也較高。在這個反應體系中,碳負離子作為親核試劑,進攻帶有正電荷的原子和離子。質子性溶劑做反應體系的溶劑,會導致體系中生成的碳負離子變回到對甲基苯乙酮,延長了反應時間。所以選用非質子溶劑有利于反應的進行,縮短了反應時間。
2 由三氟乙酸甲酯制三氟甲苯丁二酮
Anumula[14]在專利US7919633B2中以甲苯為溶劑,甲醇鈉作為縮合劑,分別采用對甲基苯乙酮與三氟乙酸甲酯、三氟乙酸乙酯、三氟乙酸異丙酯及三氟乙酸叔丁酯中的一種進行縮合反應合成三氟甲苯丁二酮,其中采用三氟乙酸甲酯方法進行逐次滴加方式進行投料,110℃回流24h,得到收率為92.2%,而三氟乙酸異丙酯作為原料則是以“一鍋混”的方式進行投料,75℃反應4h,收率僅為68.6%。
說明加料方式和反應條件對其收率影響較大。方法1中逐次滴加及高溫反應明顯提高了產物收率,該方法的優點是減少了反應過后蒸出溶劑的過程。另外由于用三氟乙酸異丙酯與對甲基苯乙酮縮合時,位阻較大,反應較為三氟乙酸甲酯困難,造成對甲基苯乙酮轉化率較低,收率低。
3 三氟甲苯丁二酮合成路線及機理分析
目前主要采用對甲基苯乙酮和三氟乙酸乙酯(甲酯、異丙酯)為原料通過Claisen酯縮合反應得到。其合成路線如圖2:
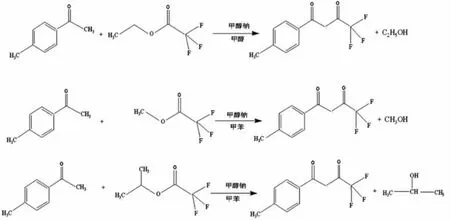
圖2 三氟甲苯丁二酮合成路線
其反應機理如圖3所示:
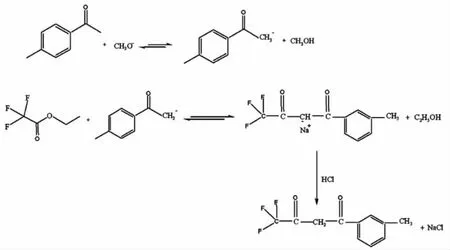
圖3 三氟甲苯丁二酮合成反應機理
4 展望
本文介紹近年來合成抗關節炎藥中間體三氟甲苯丁二酮合成工藝國內外取得的一些研究成果,三氟甲苯丁二酮合成工藝已經日漸成熟,但是作為中間體直接用于下一步塞來昔布合成的較多,中間提純的較少,并且縮合劑甲醇鈉,乙醇鈉遇水易分解,所以要求溶劑含水率極低,否則會影響縮合劑使用效果。對三氟甲苯丁二酮制備的研究主要是針對不同的反應溶劑路線來展開,其中研究較多較成熟的為甲醇、甲苯兩種溶劑,目前工藝主要存在產品純度不高、對設備要求高、原料來源不易、成本高、耗能較高及環境污染嚴重等缺點。特別是在環保形勢日益嚴峻的今天;在國外跨國公司紛紛在國內設立合資或獨資工廠;在國際金融危機下導致的原料價格上漲、產品價格及訂單下降的新形勢下,開發新的高收率,低成本,環境友好的合成路線就顯得尤為重要。
