莪術醇原藥高效液相色譜分析方法研究
2021-04-12 10:59:44鄭楊,李巖,劉博,周芹
農藥科學與管理 2021年3期
鄭 楊,李 巖,劉 博,周 芹
(1.黑龍江省植檢植保站,黑龍江 哈爾濱 150090;2.中國農業大學植物保護學院,北京 100083;3.黑龍江大學農學院,黑龍江 哈爾濱 150080)
1 前言
目前國內對莪術醇原藥分析方法尚未見報道。本文采用高效液相色譜法對莪術醇進行定性和定量分析。該方法操作經濟、快速、簡便、準確,分離效果好,準確度和精密度均能達到定量分析的要求,適用于農藥產品質量的檢測分析。
2 試驗部分
2.1 試劑和溶液 乙腈:色譜純;甲酸:分析純;超純水(電阻率18.2MΩ·cm,25℃);莪術醇標樣:99.1%;試樣:莪術醇原藥。
2.2 儀器 高效液相色譜儀:Agilent1260,具有二級管陣列檢測器和自動進樣器,定量環20μL;色譜工作站;Millipore超純水制備系統;色譜柱:250mm×4.6mm(i.d)不銹鋼柱,內裝Zorbax SB-C18、5μm填充物;過濾器:濾膜孔徑約0.45um。
2.3 液相色譜操作條件 流動相:乙腈:0.1%甲酸水溶液=85:15;流速:1.0mL/min;柱溫:30℃;檢測波長:210nm;進樣體積:5.0uL;保留時間:莪術醇5.3min。
2.4 測定步驟
2.4.1 標樣溶液的配制 準確稱取莪術醇標樣0.031g(精確至0.2mg),置于100mL容量瓶中,用乙腈超聲溶解并稀釋至刻度,靜置備用。
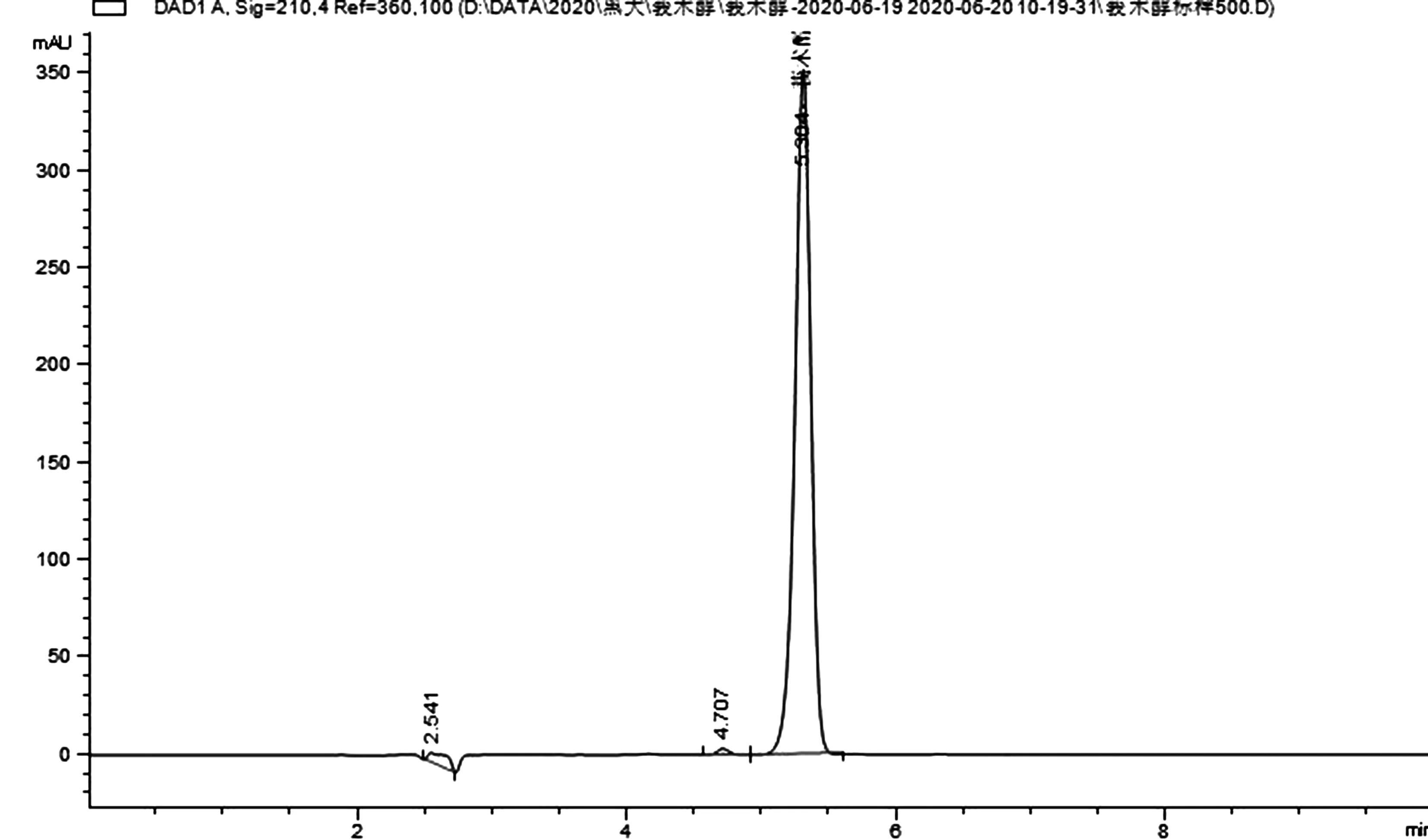
圖1 莪術醇標品的液相色譜圖
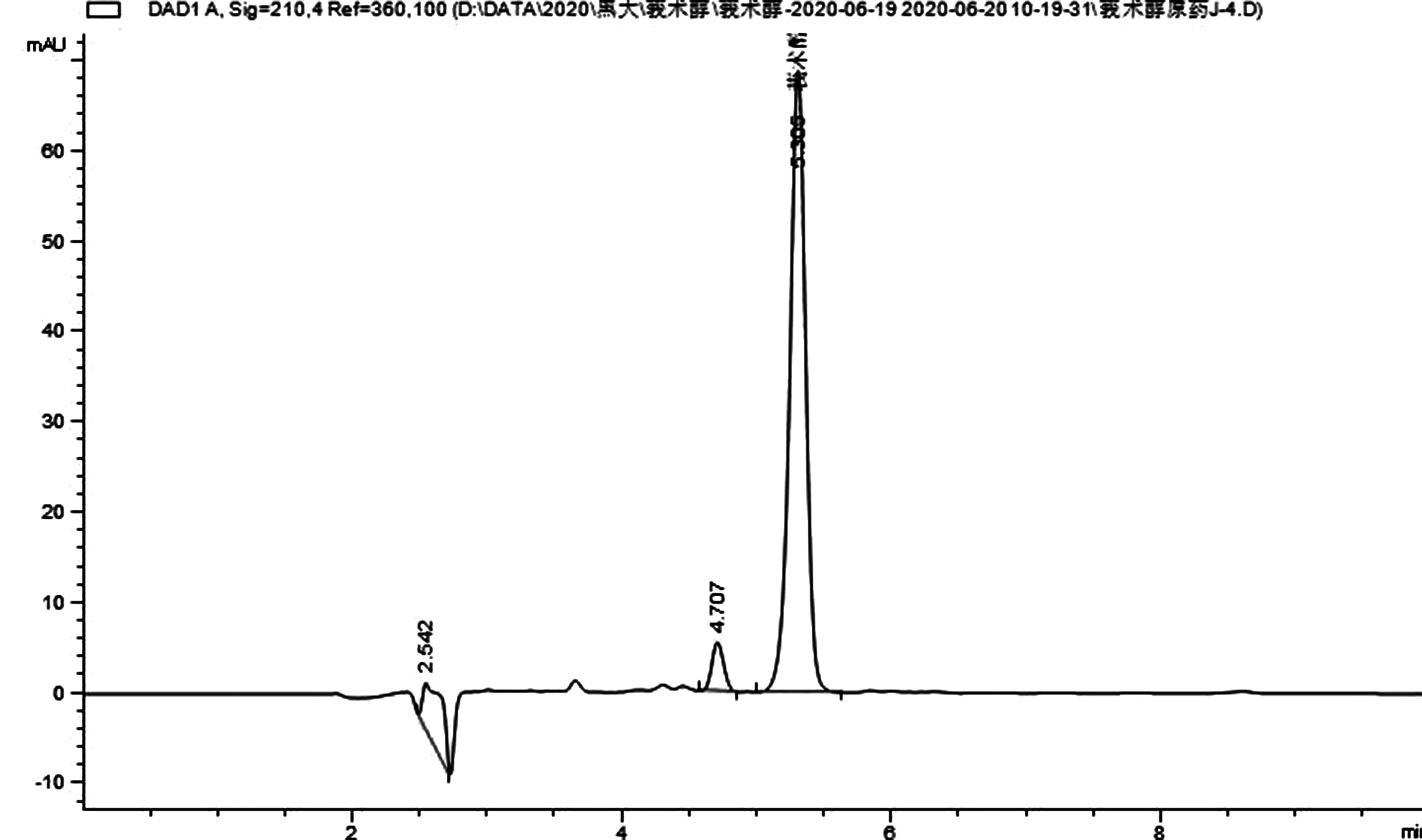
圖2 莪術醇92%原藥樣品的液相色譜圖
2.4.2 試樣溶液的配制 稱取試樣0.026g(精確至0.2mg),置于100mL容量瓶中,用乙腈溶液超聲溶解并稀釋至刻度,靜置備用。
2.4.3 測定 在上述操作條件下,待儀器基線穩定后,連續注入數針標樣溶液,待各針相對響應值變化﹤1.5%時,按照標樣溶液、試樣溶液、試樣溶液、標樣溶液的順序進樣測定。
2.4.4 計算 將測得的2針試樣溶液及試樣溶液前后2針標樣溶液中莪術醇的峰面積值分別進行平均,按下式計算其質量分數:
式中:
A1—標樣溶液中莪術醇峰面積的平均值;
A2—試樣溶液中莪術醇峰面積的平均值;
m1—標樣的質量,g;
m2—試樣的質量,g;
P —標樣中莪術醇的質量分數,%。
3 結果分析
3.1 色譜條件的選擇 通過Agilent ZORBAX 高效液相色譜儀的光譜數據采集功能,獲得莪術醇的紫外吸收圖(圖3),從圖中可以看到在吸收波長190~400nm區域內,莪術醇的最大吸收波長在200nm附近,由于吸收波長200nm附近溶劑干擾比較明顯,因此將檢測波長定為210nm。
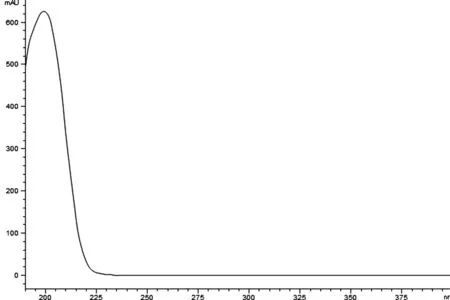
圖3 莪術醇紫外吸收譜圖
依據莪術醇的理化性質,選用乙腈和甲酸水溶液作為流動相,用乙腈溶解樣品,有效成分與雜質能得到很好分離,峰形對稱,基線平穩,分離效果好。
3.2 分析方法的線性相關性試驗 準確稱取莪術醇標樣0.033g(精確至0.2mg),置于50mL容量瓶中,用乙腈超聲溶解并稀釋至刻度,靜止備用。用移液管準確移取上述標準溶液2.0、4.0、6.0、8.0、10.0mL于10mL容量瓶中,用乙腈定容至刻度搖勻后備用。按上述條件進行測定,以進樣濃度為橫坐標,峰面積為縱坐標,繪制標準曲線,得線性方程為y莪術醇=9 632.27x-5.39,相關系數為0.999 9。
3.3 方法精密度的測定 在上述色譜操作條件下對同一樣品平行測定5次莪術醇的標準偏差為0.10;變異系數為0.11%(表1)。

表1 分析方法的精密度試驗結果
3.4 方法準確度的測定 按照配方比例要求,將除原藥以外的所有填料助劑混勻,作制劑空白,按照制劑標稱值在制劑空白中添加有效成分,合成5個制劑樣品,在上述色譜操作條件下進行分析,測得莪術醇的平均加標回收率為99.93%(表2)。
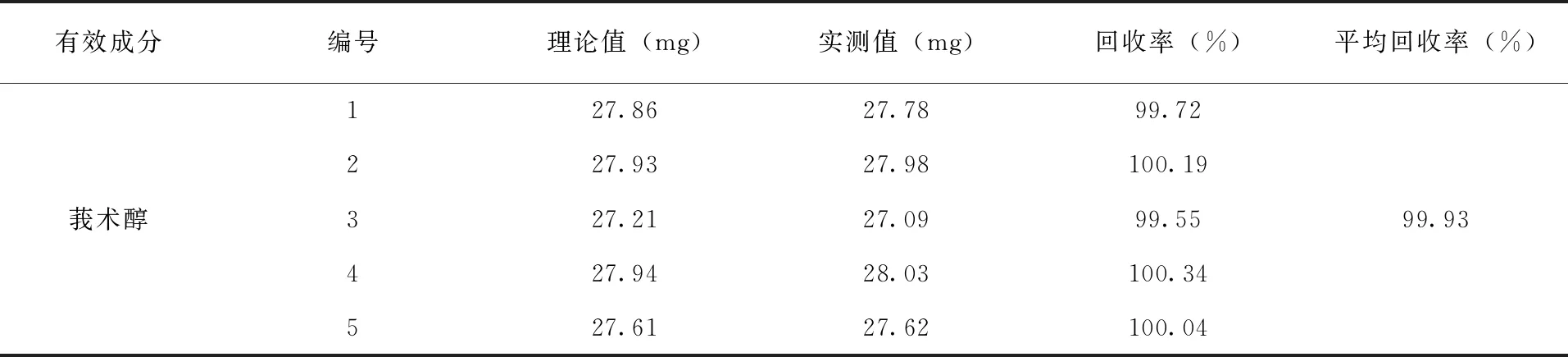
表2 分析方法的準確度試驗結果
3.5 方法特異性 對樣品組分色譜峰進行峰純度試驗,通過工作站軟件計算匹配度,峰純度因子為999.89,特異性符合要求。
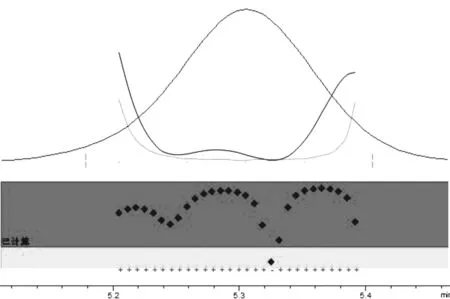
圖4 莪術醇純度
4 結論
本文建立了檢測莪術醇原藥中有效成分質量分數的高效液相色譜分析方法。試驗結果表明,莪術醇在測試濃度范圍內線性關系良好,有較高的準確度和精密度,具有簡便、快速、分離效果好的優點,是一種可行的分析方法。
