基于X射線吸收譜研究鑭系錒系化合物成鍵共價性
2021-04-20 03:10:40張宇生孫濤祥
核化學與放射化學 2021年2期
張宇生,孫濤祥
清華大學 核能與新能源技術研究院,北京 100084
化學鍵是化學科學中最基本也是最重要的概念之一,與物質的化學性質息息相關[1]。化學鍵大致上可分為兩大類:離子鍵和共價鍵,其中離子鍵的本質是陰陽離子間的電荷作用,而共價鍵則是原子間電子云重疊而形成的相互作用;對共價鍵的研究一直伴隨著化學理論的發展。自1897年Thomson發現電子,各種研究共價鍵的理論包括酸堿電子理論(Lewis theory)、價鍵理論(valance bond theory)、雜化軌道理論(hybrid orbital theory)、分子軌道理論(molecular orbital theory)、晶體場理論(crystal field theory)以及配位場理論(ligand field theory)等的發展,使得化學家對共價鍵的認識逐漸深入[2]。對成鍵理論研究的不斷深入,也大大促進了化學尤其是配位化學的發展。在元素周期表中,過渡金屬元素(d區元素)可與非金屬元素(p區元素)形成配合物,對過渡金屬配合物成鍵共價性的研究一直是配位化學研究的重點科學問題。通過分子軌道理論與配位場理論,化學家已經對過渡金屬的d軌道與配體的p軌道的成鍵行為有了比較清晰的認識[3]。
在元素周期表中,4f區過渡元素(鑭系元素)和5f區過渡元素(錒系元素)統稱為f區元素,它們的很多性質與過渡金屬類似,但同時其又表現出許多特異的性質,如鑭系收縮、錒系收縮以及錒系元素氧化態的復雜性等。目前,鑭系元素已被廣泛應用于催化、醫療設備、磁性材料、高性能合金、能源以及國防相關領域等工業生產中,而錒系元素則與能源、環境、國家安全等密切相關。對鑭系與錒系元素物理化學性質的研究已經經歷了一個多世紀,但其成果與其他元素相比仍然滯后,鑭系與錒系配位化學目前仍是相關領域的化學工作者們努力研究的主題[4-5]。究其原因,主要是鑭系與錒系元素的f軌道和d軌道均可以參與成鍵,令成鍵模式比單純d軌道成鍵的過渡金屬元素更為復雜。對于鑭系元素,研究者對4f軌道參與成鍵的程度存在一定的分歧。前期的計算表明4f軌道不會明顯擴展到芯軌道之外,認為4f軌道是定域的,所以鑭系元素化合物4f軌道不參與成鍵。然而,最近的理論和光譜研究發現4f軌道在化學鍵中扮演重要角色。與鑭系元素相比,錒系元素的5f和6d軌道具有更寬的徑向分布,可以推測錒系元素5f、6d軌道與配體軌道混合作用比鑭系元素4f、5d軌道與配體軌道混合作用強,即參與共價程度更高。同時受相對論效應和強電子相關效應影響,令其化合物成鍵模式更為復雜。5f軌道定域程度以及5f和6d軌道對共價相對貢獻成為研究者爭論的核心問題。由于4f、5f元素電子態的獨特性,研究鑭系和錒系元素共價的實驗和計算極具挑戰性,但更好地定義f軌道和d軌道在鑭系和錒系化學成鍵中所起的作用以及定量研究f、d軌道對共價的貢獻可以實質性地推進f區元素物理與化學領域的科學進展。
對f軌道和d軌道參與成鍵行為,即成鍵共價性的研究,實驗上主要是通過分子光譜學進行研究,即通過電子態之間的躍遷行為研究軌道參與成鍵行為。圖1簡單示意了常見的光譜對應的電子躍遷,包括X射線光電子能譜/紫外光電子能譜、紫外-可見吸收光譜、熒光光譜等常見的表征手段已經用于鑭系與錒系化合物的配位化學研究。除圖1所示的光譜手段,核磁共振、電子順磁共振等手段也可以反映配合物中的成鍵行為。近年來,隨著同步輻射光源的快速發展,X射線吸收譜(X-ray absorption spectroscopy, XAS),尤其是配體的K邊吸收譜,結合量子化學(主要是密度泛函理論)計算,可獲得分子軌道能級、分子軌道成分等電子結構信息,成為研究配合物中金屬與配體鍵共價的有效方法[6]。這種方法目前已經成功用于d區金屬元素-配體化學鍵共價的研究,但是對f區元素化合物成鍵的共價性研究的應用還處在發展階段。本文概述了配體X射線吸收譜與密度泛函理論兩者結合來研究元素共價的理論背景與分析方法,并分析了近些年應用此技術方法對鑭系錒系元素共價的一些研究,并進行總結與展望。

圖1 典型分子光譜所對應的電子躍遷示意圖
1 X射線吸收譜簡介
X射線吸收譜早在20世紀20年代就已經發現,但是對吸收邊前后不同能量段精細結構產生機理的研究不清楚以及沒有合適的X射線源,限制了其發展。同步輻射光源提供了強度高、亮度大、可極化、準直性好、可調諧的軟硬X射線光束,使得發展緩慢的X射線吸收譜的研究獲得了新的生命力,由此形成了一種新的電子和幾何結構研究的方法。
X射線射到樣品上與物質發生作用,穿過樣品光強度減弱,吸收系數與光強度及樣品厚度關系為:
(1)
式中:μ(E)為吸收系數,I為透射光強度,I0為入射光強度,x為樣品厚度。測量的X射線吸收隨入射光能量變化的曲線為X射線吸收譜,如圖2所示。

圖2 典型X射線吸收譜示意圖
吸收系數在整個能量波段范圍內不是單調變化的,在某些位置發生吸收突躍,此時光子能量正好對應于物質中某元素內殼層電子的束縛能,這位置稱為元素的吸收邊。根據吸收邊位置,X射線吸收譜可分為X射線吸收近邊結構(XANES)和擴展X射線吸收精細結構(EXAFS),而XANES又可以分為邊前區(pre-edge)、吸收邊(edge)和近邊區(near-edge)三部分。EXAFS部分可進行傅里葉變換,轉換為k空間和R空間,進行數據擬合后,可得到吸收光原子周圍配位原子的種類和配位數等平面結構信息;而XANES可提供吸收光原子的有效電荷、配位對稱性、化學鍵類型以及近鄰配位原子的原子種類、配位數、位置等立體結構信息。對于分子體系的X射線吸收譜,基本可以用短程有序理論來解釋,即原子直接受環境的影響,只對短程結構變化敏感,不涉及長程有序問題。EXAFS可以認為是外部X射線激發的光電子被周圍原子反散射,與出射波相干涉而引起出射光電子終態波函數變化,從而導致吸收譜產生連續的震蕩。目前已經發展了單散射理論和多重散射理論來對EXAFS進行解釋。XANES則與分子中的電子躍遷有關。當入射光子的能量足以克服對內層原子的吸引能,把它提升到高能處未占據軌道時,就會發生吸收現象。XANES可以用分子軌道理論、配位場理論、能帶理論以及多重散射理論等進行解釋。其中,分子軌道理論可有效解釋X射線吸收譜的邊前區躍遷,邊前區特征峰與內層電子向未占據或單占據的分子軌道躍遷密切相關[7-8]。
根據分子軌道理論,在金屬化合物中,金屬原子的d和f軌道(φM)可以與配體原子的p軌道(φL)形成共價鍵。成鍵軌道(占據軌道)可表示為:
Ψ=(1+2λSML+λ2)-1/2(φM+λφL)
(2)
相應的,反鍵軌道(非占據軌道)可表示為:
Ψ*=(1-2λSML+λ2)-1/2(λφM-φL)
(3)
式中:λ為軌道混合系數,SML為軌道重疊積分。當λ為1時,可以認為金屬與配體完全共價成鍵;而當λ為0時,則認為二者形成離子鍵。軌道混合系數λ可表示為:
(4)

配合物的X射線吸收譜可以反映配合物中金屬和配體的成鍵共價性。在對金屬配合物進行XAS研究時,一般研究金屬原子的L邊和M邊。金屬的L邊XAS探測從金屬的2p軌道到含有金屬d軌道成分的未占據態的分子軌道的電偶極允許躍遷,由于自旋-軌道耦合效應,分為L3和L2邊。對于存在不同配體環境情況下,躍遷強度反映了整個配體環境的凈效應下電子占有率以及電子組態,雖然不能直接探測某個特定的配體-金屬鍵的共價,但是提供了一種特別敏感和精確的方法來直接探測金屬d軌道對單占據或未占據分子軌道的貢獻,間接地對金屬-配體鍵的共價進行評估。金屬的M邊是金屬的3d軌道到含有金屬f或p軌道成分單占據或未占據態軌道的躍遷,它提供了一個獨特的探測金屬化合物中f電子占據和電子結構的實驗方法。同樣由于自旋軌道耦合效應,分為M5和M4邊。金屬的M3,2邊是3p→nd的躍遷,相比于L3,2邊,金屬M3,2邊激發能量低,因此提供了更好的分辨率,對電子態更加敏感,更有助于研究d軌道成鍵共價性。
金屬L邊和M邊X射線吸收譜的躍遷強度可以反映金屬與配體軌道混合,但是此躍遷強度反映出整個配體環境的凈效應,不可以直接探測出特定的金屬-配體鍵的共價程度。由于配體的p軌道參與成鍵,因此如果研究配體的K邊,則可描述為電子從配體的1s軌道躍遷到含p成分的分子軌道(如圖3所示)。因為1s軌道定域在配體上并且電子從s軌道躍遷到p軌道為電偶極允許躍遷,躍遷強度可以定量地探測出單占據或未占據分子軌道中含有配體p成分。所以,配體的K邊XAS可以直接定量地探測出金屬-配體鍵的共價程度。相比于d區金屬,鑭系和錒系元素共價成鍵存在很大爭議,采用配體K邊XAS并結合金屬的L邊和M邊XAS分析得到的研究化合物共價的XANES光譜數據更具有說服力。

圖3 配體K邊躍遷示意圖
常見的配體原子的K邊的能量一般較低,主要有C(284.2 eV)、N(409.9 eV)、O(543.1 eV)、F(696.7 eV)、S(2 472 eV)、Cl(2 822 eV)等。對于這些配體K邊XAS的檢測,需要在同步輻射光源的軟X射線線站或中能X射線線站完成。目前國內運行的同步輻射光源有北京同步輻射裝置(Beijing Synchrotron Radiation Facility,簡稱BSRF)、位于合肥的國家同步輻射實驗室的專用同步輻射光源(簡稱合肥光源)以及上海光源(Shanghai Synchrotron Radiation Facility,簡稱SSRF),三個光源的上百個實驗站提供從紅外光到硬X射線的高強度、高亮度、高準直性等特性的同步輻射光,成為科學前沿領域研究和高技術開發不可或缺的實驗平臺,可提供X射線吸收譜的測試。例如,北京同步輻射光源的中能線站和軟線線站可提供從100~3 000 eV范圍的吸收譜實驗。XAS的檢測方法主要有全電子產額(total electron yield, TEY)、熒光產額(fluorescence yield, FY)、掃描透射X射線顯微(scanning transition X-ray microscope, STXM)等方法。其中全電子產額方法是檢測X射線吸收所產生的Auger電子和光電子的總和,FY是檢測熒光產額。在TEY和FY方法中,樣品表面污染、自吸收以及飽和效應等因素會降低XAS峰的強度,而STXM在一定程度上可以克服此困難,得到相對精準的配體K邊XAS數據。同步輻射光源測得的XAS數據,可以用Demeter等相關的專業軟件進行處理,得到相關的物理化學信息[9]。
2 密度泛函理論簡介
X射線吸收譜的邊前峰的峰位置與峰強度,與分子的軌道能級以及軌道成分密切相關。因此,對X射線吸收譜的解釋,尤其是對邊前峰的理論解釋,離不開現代量子化學計算手段。量子化學的理論源于量子力學,其核心問題是求解原子-分子體系的Schrodinger方程。量子化學的計算方法主要包括波函數理論(wave function theory, WFT)和密度泛函理論(density functional theory, DFT)。波函數方法的計算量通常較大,簡單的Hartree-Fock(HF)方法計算量與基函數數目的3~4次方成正比,后HF方法計算量則呈數量級增長。因此,在電子數目較多的鑭系、錒系化合物的計算中,波函數理論方法不是一個高效方法。在密度泛函理論中,體系的能量和其他性質可以表達為電子密度ρ(r)的泛函,因此計算時只需對電子密度分布函數進行變分,極大地減少了計算量,成為量子化學計算的主流方法之一[10-13]。
密度泛函理論的基礎是Hohenberg和Kohn提出的電子密度和體系能量相關的兩個定理。HK第一定理指出體系的基態電子密度可完全確定體系的所有性質,因此體系的總能量可以寫成密度函數的泛函形式。HK第二定理指出體系的總能量服從變分原理。雖然HK定理給出了DFT的嚴格理論證明,但沒有給出泛函的具體形式,也沒有提供由電子密度計算基態性質的實際方法。Kohn和Sham在HK定理的基礎上,引入非相互作用的參考系,將體系的動能和庫倫能從泛函中分離出來,再經過近似處理得到可以應用于實際計算的Kohn-Sham方程:
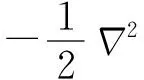
在重元素原子核附近的內層電子運動速率很快,相對論效應顯著,導致重元素在原子基態、電離勢、電子親合勢和原子半徑等方面有不同于輕元素的特點,并影響其化學性質。因此鑭系和錒系元素理論化學計算時需要考慮相對論效應[14]。密度泛函理論中對相對論的考慮主要有全電子法和有效核實勢法。核實勢法(effective core potentials, ECPs)的主要物理思想是將原子的內層電子連同原子核構成一個核實,核實對外層電子的作用以某種合適的有效核實勢來表達,與全電子法相比可大大降低計算量。ECPs主要有模型勢(model potentials, MPs)和贗勢(pseudo potentials, PPs)兩個分支,后者的配套基組通常更小,因此計算量更低,應用更廣泛。
光的吸收和發射過程涉及電子從一個能級向另一個能級的躍遷。Kohn-Sham方程雖然在理論上包含了電子相關作用,但形式上是單電子方程,原則上只適用于體系非簡并基態電子結構的計算。基于線性響應理論的含時密度泛函方法在密度泛函理論框架內,解決了激發態計算問題,對激發能的計算誤差一般為0.1~1.0 eV,已經成為計算大分子體系、鑭系和錒系體系激發態性質最知名的方法。
3 鑭系和錒系化合物成鍵共價性研究
XAS結合DFT的方法已經成功用于研究過渡金屬配合物的成鍵共價性[15-16]。例如,此方法可以用于研究金屬硫蛋白中金屬離子Cu(Ⅱ)、Fe(Ⅱ)等與硫原子的成鍵行為,揭示金屬的d軌道與硫的p軌道的混合,以解釋相應蛋白質的功能[17]。如前所述,相比于普通過渡金屬元素,鑭系和錒系元素可同時以f和d軌道參與成鍵,成鍵行為更為復雜,導致其XAS譜圖的邊前峰也更為復雜。
在理論計算方面,鑭系、錒系元素有著極大的體系尺寸和電子數、電子相關作用強以及電子態能量簡并度高,綜合考慮計算量與精確度,采用DFT方法并結合合適的交換相關泛函進行計算是合適的,DFT和TDDFT已經成為目前應用量子化學的主流方法。采用DFT計算出化合物基態下分子軌道能量、分子軌道組成以及詳細的電子結構信息,對于理解分子的虛電子態和XAS光譜特征峰指認具有重要的理論支撐作用。應用TDDFT計算出電子躍遷的激發能和電子激發的振子強度對光譜進行模擬,并對光譜解釋提供理論指導。
XAS與DFT結合是目前探測元素-配體化學鍵共價最直接有效的方法。與此同時,XAS與DFT計算的結合也可以用來驗證和改進理論計算方法提高在鑭、錒系元素方面的理論計算水平。目前已有的文獻[18-33]報道中,研究比較多的是含C、N、O、Cl等配體原子的鑭系和錒系化合物。
3.1 碳K邊XAS
金屬-碳鍵(M-C)的化學性質在金屬有機化學中發揮著主導作用,但是實驗上對M-C成鍵的共價性研究極少。為了對比d區元素與f區元素金屬-碳成鍵的共價性,Minasian等[18,34]采用碳的K邊XAS與DFT相結合的方法先后對(C5H5)2MCl2(M=Ti、Zr、Hf)以及(C8H8)2An(An=Th、U)進行了研究。


圖4 金屬Ti、Zr、Hf的d軌道與的π軌道作用示意圖[34]

圖5 (C5H5)2MCl2(M=Ti、Zr、Hf)分子的碳的K邊XAS數據及分峰擬合結果[34]


圖6 (C8H8)2An(An=Th、U)中金屬的5f和6d軌道與的π軌道的相互作用[18]


圖7 (C8H8)2An(An=Th、U)中碳K邊XAS的實驗結果與TDDFT計算結果對比[18]
3.2 氮K邊XAS
與硬原子(如氧)配體相比,含氮軟配體對鑭系和錒系化合物具有更強的分離能力。對含氮軟配體與錒系化合物的電子結構性質的研究,有助于深入認識含氮軟配體的配位能力,從而進一步認識其分離能力。2,6-二(2-苯并咪唑)吡啶(2,6-bis(2-benzimidazyl)pyridine, H2BBP)分子具有一個嘧啶環和兩個咪唑環,可以以氮原子與金屬離子配位。Pemmaraju等[20]采用氮K邊XAS和理論模擬研究了H2BBP與鈾酰化合物的電子結構。鈾酰離子與H2BBP配位后,其赤道面上的空位還可以與Cl-配位,形成UO2(H2BBP)Cl2。在這個化合物中,存在三種化學環境的氮原子,因此氮K邊XAS譜圖上出現多個峰。如圖8[20]所示,單獨檢測H2BBP的氮K邊XAS時出現四個邊前峰,但化合物UO2(H2BBP)Cl2的XAS譜圖中主要有三個邊前峰Ⅰ、Ⅱ、Ⅳ,未發現峰Ⅲ。對不同化學環境的氮原子分別計算1s至空軌道的躍遷,發現峰Ⅰ主要由位于中心吡啶位點N2的1s到π*躍遷貢獻,咪唑環上N1位點的1s到π*躍遷對峰Ⅰ也有貢獻;峰Ⅱ主要來源于咪唑環上N1位點的1s到π*躍遷和N4位點上1s到π*躍遷;峰Ⅳ來源于N4位點的1s到π*躍遷以及N1位點的1s到π*躍遷。相比于配體,配合物中的邊前峰均右移約0.2 eV,這是由于發生配體向金屬的電荷轉移,氮原子的1s能級更低。

圖8 UO2(H2BBP)Cl2分子中的配位模式(左)、TDDFT計算H2BBP和UO2(H2BBP)Cl2中不同N原子的激發所得的理論氮K邊XAS譜圖(中)以及H2BBP上不同位點的N原子與相互作用的最低能量的孔穴激發態的分子軌道圖(右)[20]
3.3 氧K邊XAS
了解鑭系和錒系氧化物中的金屬的f、d軌道與氧的2p價軌道的混合,為鑭系和錒系的氧化物的成鍵和電子結構建立一個定量的模型,不論在實驗方面還是在理論方面都有相當大的挑戰性。鑭系的氧化物主要為Ln2O3,部分鑭系如Ce、Pr、Tb可形成LnO2。對錒系元素U,UO2是一種常用的核燃料,而U3O8則是最穩定的氧化物。
Altman等[21]應用氧K邊XAS來研究除Pm以外的所有的Ln2O3的電子結構以及軌道混合的變化。所有的Ln2O3的氧K邊XAS譜圖中均有三個邊前峰,由于Lu(Ⅲ)的4f軌道是全占據的,因此這三個邊前峰對應的躍遷終態應當對應于Ln的5d和6p軌道與O的2p軌道的混合:即5dπ、6p、5dσ。進一步對躍遷強度進行分析,1s→5dσ的躍遷貢獻最大,說明O的2p與Ln的5d軌道的σ成鍵對Ln-O成鍵的貢獻最大。4f 軌道的能量高低和占有率會影響 Ln的5d軌道和O的2p軌道混合的程度,導致電子結構相似的化合物有著不同的成鍵模式。
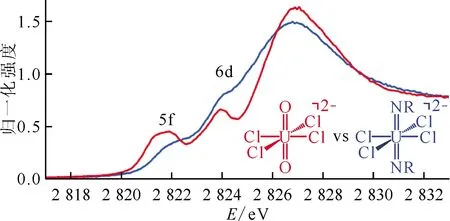
Minasian等[22]通過氧K邊XAS定量地研究LnO2(Ln=Ce、Pr、Tb)中O的2p軌道與Ln的4f、5d軌道混合程度。LnO2的氧K邊XAS譜圖示于圖9[22]。由總的態密度和O的2p軌道部分態密度計算可知,XAS譜圖中高能區域的峰與O的2p軌道和Ln的5d軌道混合相關,低能部分與O的2p軌道和Ln的4f軌道混合相關。計算結果顯示,O的2p軌道與Ln的5d軌道形成σ型和π型鍵,和Ln的4f軌道形成σ鍵,而僅有PrO2觀察到4f軌道與O的2p軌道形成π鍵。實驗與計算結果顯示O的2p軌道與Ln的4f軌道混合程度為TbO2 圖9 LnO2中Ln的4f軌道與O的2p軌道的σ成鍵示意圖(左)和LnO2的氧K邊XAS譜圖(右)[22] Jollet等[23]采用氧K邊XAS和理論計算來研究UO2的電子結構及U-O成鍵的共價性,并與CeO2的電子結構及成鍵共價性進行對比。UO2的XAS譜中的邊前鋒由O的2p與U的5f和6d軌道混合而產生,結合態密度計算可以認為,前兩個峰為電子從O的1s軌道到O的2p軌道與U的5f軌道混合的分子軌道躍遷,后兩個特征峰為電子從O的1s軌道到O的2p軌道與U的6d軌道混合的分子軌道躍遷。 Wen等[24]采用氧K邊XAS來研究U3O8中U-O鍵O的2p軌道與U的5f、6d軌道混合作用,采用掠入射熒光產額、熒光產額和透射三種探測方式進行數據采集。由于飽和效應影響,透射模式到熒光和掠入射熒光模式測得的總的峰強度逐漸減小。在XAS中存在兩個邊前峰,分別位于530.8 eV和536.6 eV附近。態密度計算可以確認U的5f空帶能量低于6d空帶能量,并且U(Ⅵ)的5f、6d空帶能量低于U(Ⅴ)的5f、6d空帶能量。O的2p態密度分為兩個區域:第一區域為O的2p軌道與U的5f軌道混合,第二區域為O的2p軌道與U的6d軌道混合,從而在XAS譜圖中第一個峰對應于1s→5f,第二個峰對應于1s→6d。 圖10 中氯的3p與金屬的f和d軌道混合形成的反鍵軌道示意圖[30] 圖11 的氯K邊XAS結果[31] 圖12 的氯K邊XAS譜圖以及軌道能級躍遷示意圖[32] 圖13 的反鍵軌道作用圖[33] 圖14 與的氯K邊XAS譜圖對比[33] 配體的K邊XAS結合密度泛函理論計算是目前定量研究鑭系和錒系化合物成鍵共價性的最直接有效的實驗方法,譜的邊前峰位置反映分子的軌道能級,而峰的強度直接反應軌道混合的程度;理論計算結果為光譜的理論解釋提供指導,二者相結合可探索鑭系和錒系化合物復雜的電子結構,揭示鑭系和錒系化合物中f和d軌道的成鍵行為。盡管目前研究的體系有限,主要集中在鑭系和錒系的氧化物和氯化物,但是初步的結果已經表明鑭系的4f和錒系的5f軌道在共價成鍵中均扮演重要的角色;相對來講,更彌散的錒系的5f軌道參與成鍵的程度更高。一些非常重要的結果讓科研工作者對鑭系和錒系化合物的電子結構有了更深入的認識,如錒系5f軌道參與成鍵的能量簡并驅動效應。 盡管X射線吸收譜與密度泛函理論計算結合的方法已經對鑭系和錒系化合物電子結構獲得了很多有意義的結果,但對相關問題的研究仍非常具有挑戰性,主要表現在以下幾個方面。 (1)目前對錒系化合物的研究主要集中在Th和U兩種元素,對超鈾元素的研究非常少。超鈾元素的放射性導致對其實驗上的合成相對困難,更導致對化合物進行XAS數據的采集更加困難。目前國內同步輻射光源對放射性元素的測試沒有建立明確的操作條例,并且放射性樣品的運輸存在同樣的問題,這些均限制了對超鈾元素化合物的研究。實際上,在防護措施完善的情況下,完全可實現超鈾元素化合物的合成、運輸、測試等一系列流程。美國在這方面則已經有完善的流程,可以進行超鈾元素的運輸、同步輻射測試等。我國目前亟需的是相關部門在相關方面制定相關法律法規和操作規程。 (2)目前對XAS實驗譜數據的解釋主要是通過密度泛函理論計算進行。但是,密度泛函理論的基礎仍為單電子近似,對含f電子的強關聯體系的計算存在一定的缺陷。更重要的是,XAS方法探測的是未占據軌道,而單電子近似的相關理論對未占據軌道的能級和軌道成分計算仍存在問題。因此,在綜合考慮計算量與精確度的前提下,發展新的理論計算方法尤為必要。 (3)基于大科學裝置發展新的表征手段,仍是目前化學理論研究的關鍵推動力。基于同步輻射光源的更為先進的線站技術,如X射線發射譜(X-ray emission spectroscopy, XES)、X射線拉曼譜(X-ray Raman spectroscopy, XRS)等的發展,將有助于更加深入地表征鑭系和錒系化合物的電子結構;新一代X射線自由電子激光的建成,將會使相關的表征方法具有更高的時間空間分辨率,因此獲得更精細的電子結構。實驗技術以及理論計算方法的協同發展,勢必會推動鑭系和錒系相關領域的基礎科學發展。
3.4 氯K邊XAS

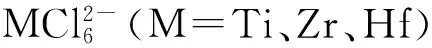



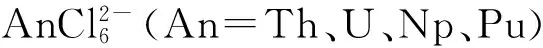





4 總結與展望
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
當代陜西(2022年5期)2022-04-19 12:10:18
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:28
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
湘潮(上半月)(2021年4期)2021-07-20 08:05:28
汕頭大學學報(自然科學版)(2020年4期)2020-12-14 07:05:00
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
汽車工程學報(2017年2期)2017-07-05 08:13:02
