腎意義的單克隆免疫球蛋白血癥病例報道并文獻復習
2021-04-23 07:58:10魏宇君李新申曼張佳佳詹曉凱湯然黃仲夏
中國全科醫學 2021年18期
魏宇君,李新,申曼,張佳佳,詹曉凱,湯然,黃仲夏
多發性骨髓瘤(MM)是一種伴單克隆免疫球蛋白(Ig)積聚的惡性漿細胞(PC)病,50%的MM患者存在腎損害。少數單克隆Ig升高的患者表現為血肌酐(Scr)升高或腎功能不全等,卻不能用MM等已有疾病來解釋。2012年國際腎病和單克隆免疫球蛋白病研究組(IKMG)提出了腎意義的單克隆免疫球蛋白血癥(MGRS)的概念。2019年IKMG提出了MGRS診斷共識[1],2020年第1版美國國立綜合癌癥網絡(NCCN)納入了MGRS的診治指南。MGRS的提出是血液內科、腎內科、腎臟病理學界的一大進步,解決了困擾臨床醫師多年的單克隆免疫球蛋白血癥(MGUS)合并腎損害的診治問題。本文通過整理分析2018年3月—2020年2月首都醫科大學附屬北京朝陽醫院西院血液與腫瘤科收治的1例MM合并腎損害和3例不同類型的MGRS患者的臨床特點和診治過程,對其腎損害進行了鑒別診斷,并結合病例資料和文獻進行系統討論,以期提高廣大臨床醫師對該類疾病的診治水平。本研究中所有患者及其家屬簽署了知情同意書,并經過首都醫科大學附屬北京朝陽醫院倫理委員會批準。
1 病例簡介
病例1,男,67歲,主因“骨痛、腰椎骨折、Scr升高10 d”于2013-06-19就診于首都醫科大學附屬北京朝陽醫院。體格檢查:強迫臥位,不能行走。實驗室檢查:Scr 487.5 μmol/L。M蛋白鑒定:血蛋白電泳顯示一高尖的M蛋白波峰沉積于α2—γ區,血免疫固定電泳IgA陽性,IgA 37.1 g/L。尿免疫固定電泳κ輕鏈陽性,尿κ輕鏈12.1 g/24 h。骨髓穿刺和骨髓活檢:克隆性PC達80%。免疫分型:異常漿細胞占32.86%,免疫分型檢查顯示表達CD38/CD56/CD138/ckappa/CD27,不表達CD19。影像學檢查提示胸骨、骨盆、腰椎(L1、L2)可見多發骨質破壞和骨折。結合患者病史及入院后檢查該患者診斷為:MM IgA-κ型,腎功能不全。患者按照硼替佐米聯合環磷酰胺和地塞米松(VCD)方案接受2個周期的化療后,M蛋白下降,IgA恢復正常,尿κ輕鏈下降為1.2 g/24 h,Scr恢復正常,根據2014年國際骨髓瘤工作組(IMWG)的療效標準患者達到部分緩解(PR)[2],患者的基本信息、疾病特征及轉歸見表1。
病例2,女,72歲,主因“心悸、下肢水腫2個月余”于2019-10-16就診于首都醫科大學附屬北京朝陽醫院。既往因心率緩慢曾植入心臟起搏器治療。血壓(BP)86/60 mm Hg(1 mm Hg=0.133 kPa),下肢凹陷性水腫,N末端腦鈉肽前體(NTpro-BNP)3 900 ng/L,肌鈣蛋白I>100 ng/L。心臟彩超:心室肥厚,室間隔厚度>16 cm。M蛋白:免疫固定電泳示尿λ輕鏈陽性,尿λ輕鏈0.5 g/24 h,尿蛋白6.3 g/24 h。骨髓活檢:克隆性PC占6.5%。腎活檢:剛果紅染色陽性,λ輕鏈限制性表達。根據IMWG和IKMG的診斷標準,臨床診斷為:系統性輕鏈淀粉樣變性(AL),心臟、腎臟受累。治療上給予VCD(硼替佐米+環磷酰胺+地塞米松)方案化療,4個化療周期后患者訴心悸癥狀較前改善,NTpro-BNP下降為500 ng/L,目前患者處于維持治療階段。患者的基本信息、疾病特征及轉歸見表1。
病例3,男,47歲,主因“骨痛、蛋白尿6個月”于2019-06-20就診于首都醫科大學附屬北京朝陽醫院門診。6個月前開始出現右側上肢和左側下肢疼痛,有時腰痛,2019-01-14就診于當地醫院骨科行多項影像學檢查未發現異常。血紅蛋白(Hb)、血鈣和Scr正常,查M蛋白:IgA-κ陽性,血IgA 12 g/L,尿κ輕鏈0.2 g/24 h,尿蛋白0.4 g/24 h。骨髓穿刺:骨髓中可見12%異常PC。就診醫院懷疑為“MM”,并行腎活檢,但診斷不明確,故來本院進一步診治。入院后首先經過北京大學第一醫院腎臟病理專家會診。1周后,腎臟病理結果為剛果紅染色陰性,κ輕鏈陽性;電鏡顯示腎小球上皮變性,可見顆粒狀沉積;骨髓系列檢查和復查M蛋白等檢查進一步除外了MM等疾病,診斷為輕鏈沉積病(LCDD)。予其VCD方案化療4個周期,目前已達到非常好的部分緩解(VGPR)。患者的基本信息、疾病特征及轉歸見表1。
病例4,女,49歲,主因“間斷乏力7年,加重伴骨痛、泡沫尿4個月余”于2017-04-24就診于北京朝陽醫院。患者兩次骨髓穿刺和骨髓活檢均顯示克隆性PC為6%,免疫固定電泳IgA-κ陽性,IgA 32.2 g/L,尿κ輕鏈4.8 g/24 h,尿蛋白5.39 g/24 h。Hb 90 g/L,Scr在參考范圍,血鈣下降,同時存在尿鈉、氯、鈣排出增多,氨基酸尿、尿糖水平升高及大量蛋白尿,低血鉀、低血鈣、低血磷和低鉀性酸中毒,未發現腦垂體瘤或內分泌激素異常。影像學檢查未發現骨損害證據。診斷考慮M蛋白相關的范可尼綜合征。追問病史,患者平日月經量多,經婦科手術切除子宮平滑肌瘤,補鐵及糾正低鉀、低鈣,患者貧血和骨痛明顯緩解。考慮患者的貧血和骨痛對癥治療后好轉,并非MM定義性終末期器官損害CRAB癥狀〔高鈣血癥(C)、腎損害(R)、貧血(A)和骨損害(B)〕,故MM診斷證據不足,不能除外MGRS,建議行腎活檢明確診斷。經北京大學第一醫院腎內科腎活檢,病理結果為剛果紅染色陰性,κ輕鏈陽性,電鏡下腎小管上皮胞質內結晶形成,考慮輕鏈近端腎小管病(LCPT)。根據IKMG關于MGRS的診斷標準,本例患者的臨床診斷為LCPT,接受了硼替佐米基礎方案治療,尿輕鏈和蛋白尿好轉,范可尼綜合征相關癥狀改善,目前仍在治療過程中,患者的基本信息、疾病特征及轉歸見表1。
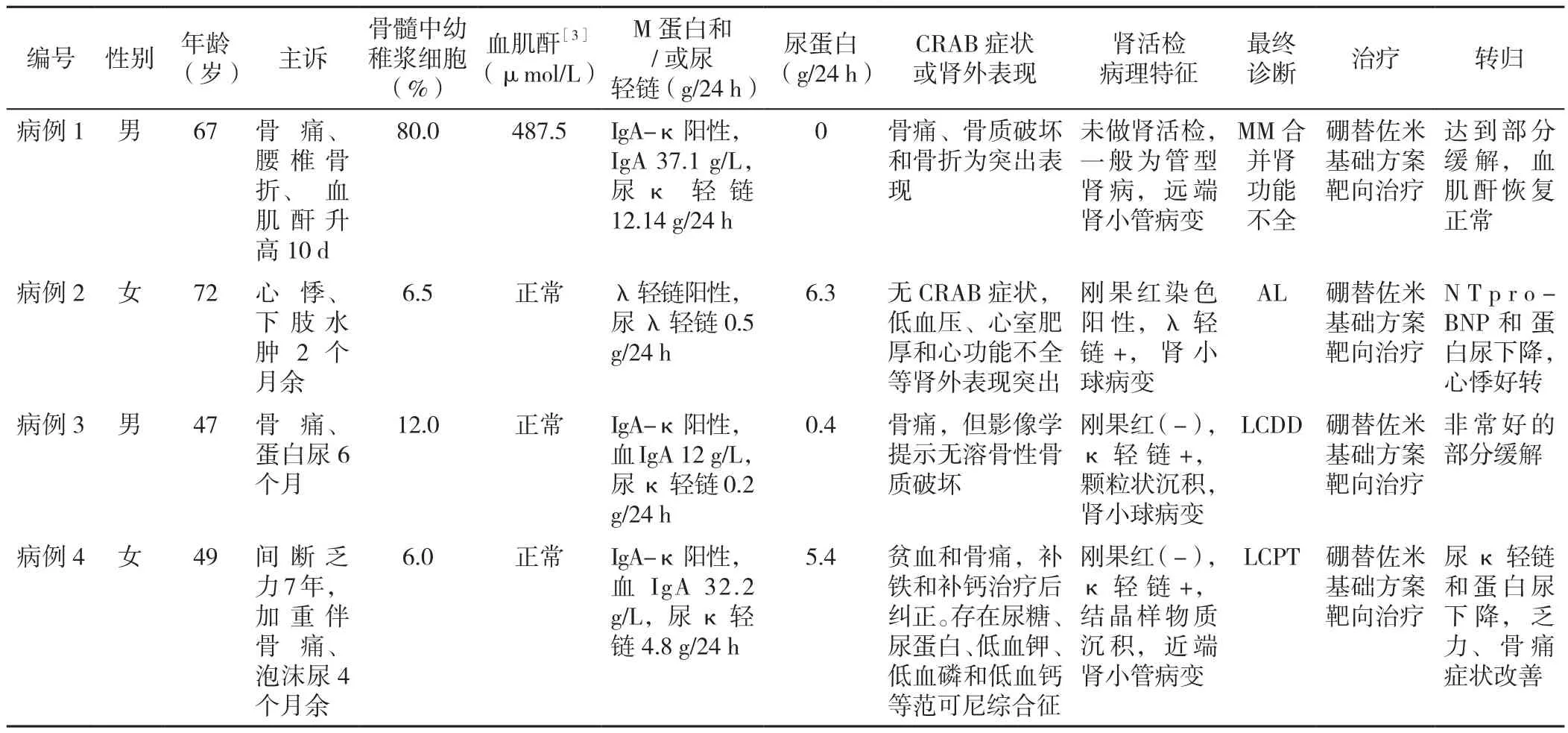
表1 4例患者一般臨床特征和腎損害情況Table 1 General clinical features and renal damage in 4 patients
2 討論
MM是一種易發生于老年人的惡性PC病,其特征是骨髓內克隆性PC超過10%,單克隆Ig積聚,并出現CRAB癥狀[4]。MM來自意義未明的MGUS或無癥狀型骨髓瘤(SMM),兩者均無CRAB癥狀,分別以每年1%和3%的速度向MM演變[5-7]。目前MM診斷依據上述IMWG的診斷標準[2],CRAB癥狀不僅是MM的定義性癥狀,也是治療的主要依據[8]。臨床觀察發現,部分患者在進展為癥狀性MM之前,已經存在腎損害,這種單克隆Ig相關的腎損害有獨特的病理特征[9],故將其從MGUS中獨立出來,由此提出了MGRS的概念[10]。2012年國際腎病和IKMG定義了MGRS,其不符合MM或淋巴瘤等已有疾病腎損害的診斷標準[11]。
2.1 MM合并腎損害 病例1表現為骨痛、腎功能損害,結合骨髓克隆性PC達80%,滿足MM的診斷標準,MM中大量Ig輕鏈損害遠端腎小管導致管型腎病是MM合并腎損害的主要病理基礎,故其腎損害是由MM引起的。此外,高鈣血癥、繼發淀粉樣變性和腫瘤局部浸潤可加重腎損害,但由于病變在腎小管,患者很少并發高血壓[12-13]。對于MM合并腎損害,早期診斷并及時給予硼替佐米基礎方案治療對患者腎功能恢復和預后至關重要[14]。本院既往對新診斷的MM合并腎功能不全的患者進行臨床分析,硼替佐米組的總體治療反應率(77.8.7%)高于非硼替佐米組(44.7%);同時,診斷時的Scr水平影響腎功能逆轉率,Scr <310 μmol/L時腎功能逆轉率(56.1%)高于Scr≥310 μmol/L(33.3%)時。腎功能在治療前3個月改善最明顯[15]。輕鏈快速下降、癥狀緩解或PR是腎功能恢復的基礎,早期發現腎損害并及時治療有利于腎功能恢復,若>45 d患者腎功能逆轉率低[16]。2007年食品藥品監督管理局(FDA)批準硼替佐米作為MM合并腎功能不全(包括透析)患者的首選治療,可逆轉40%~50% MM患者的腎功能不全[13]。
2.2 AL AL是一種危及生命的克隆性PC病,AL沉淀物多在腎小球沉積,重要臟器受累的比例為心臟82%、腎臟68%。通常伴有心功能不全或腎功能不全,腎外表現為直立性低血壓、勞累性呼吸困難、疲勞和心室壁增厚、巨舌等臨床表現[17-18]。病例2因“心悸、下肢水腫2個月余”入院,骨髓活檢:克隆性PC占6.5%。腎活檢:剛果紅染色陽性,λ輕鏈限制性表達。該患者排除MM的診斷,結合其病例資料臨床診斷為系統性AL,心臟、腎臟受累。給予VCD治療,患者心悸、心力衰竭相關指標NTpro-BNP明顯下降。AL的確診需要皮下脂肪、腎臟或其他受累器官活檢,剛果紅染色陽性為其診斷金標準[1-2]。淀粉樣蛋白在光學顯微鏡下表現呈剛果紅染色陽性,偏振顯微鏡下呈特異的蘋果綠熒光雙折射,電鏡下為直徑8~10 nm隨機排列纖維樣物質沉積,80%為λ輕鏈,需要免疫組化證實單克隆Ig輕鏈的存在,必要時需要質譜檢測,以進一步確定具體的淀粉樣變物質分子[17,19]。
既往AL患者中位生存期(OS)為1~2年,有明顯的心臟受累的患者OS僅為6個月。由于硼替佐米等靶向新藥及干細胞移植治療的開展,目前OS為平均5年以上,5%~30%患者OS已超過10年[18]。硼替佐米基礎方案是目前無此藥禁忌的AL患者標準的治療方案,心臟淀粉樣變性是影響患者生存期的主要因素,因此年輕的、有合適供者的嚴重心臟淀粉樣變性(晚期心臟受累)患者可以考慮異基因心臟,然后進行自體造血干細胞移植(ASCT),以防止疾病復發[17]。
2.3 LCDD 病例3入院后首先經過北京大學第一醫院腎臟病理專家會診。1周后,腎臟病理結果為剛果紅染色陰性,κ輕鏈陽性;電鏡顯示腎小球上皮變性,可見顆粒狀沉積;骨髓系列檢查和復查M蛋白等檢查進一步除外了MM等疾病,診斷為LCDD,給予其硼替佐米為基礎的化療方案化療4個周期后評估達到VGPR,后因疫情原因未能返院進一步診療,電話隨診患者一般狀況良好。在LCDD中,通常是κ輕鏈沉積在腎小球,在系膜區內可見輕鏈顆粒沉積。LCDD特征性臨床表現為Scr升高或腎功能不全、蛋白尿、淀粉樣變性或腎外受累[13]。
KHERA等[20]進行了以下回顧性研究,腎存活定義為從診斷為MGRS至需要腎替代療法(血液透析或腎移植等)的時間。在這項回顧性研究中,研究者分析了從2004—2017年在5個醫療中心(4個在英國,1個在愛爾蘭共和國)接受治療的41例MGRS患者資料;其中33例(80.5%)為κ輕鏈限制性表達,27例(65.9%)被診斷為LCDD。貧血33例(80.5%),BP>140/90 mm Hg 25例(61.0%)。慢性腎臟病(CKD)1~3b期14例(34.1%),CKD 4~5期為23例(56.1%),中位肌酐清除率為26 ml/min。29例(70.7%)患者接受了一線化療,其中23例患者接受了硼替佐米基礎方案化療,在21例可評估反應的患者中,≥PR者占70.8%,≥VGPR的患者無一例接受替代治療。治療24個月時,診斷為CKD2~3b期的患者顯示出其腎臟存活率為100.0%,而CKD 4~5期的患者則為80.7%(P=0.04)。隨訪48個月時,整個隊列的腎臟存活率為80.3%,分析認為某些MGRS患者的較差生存與診斷延遲有關。
2.4 LCPT 病例4主因“間斷乏力7年,加重伴骨痛、泡沫尿4個月余”入院。結合患者病史及入院后相關檢查,其不符合MM的診斷標準,通過查閱文獻,不能除外MGRS,建議行腎活檢進一步明確診斷。后經北京大學第一醫院腎內科腎活檢,確診LCPT。LCPT是以單克隆Ig輕鏈沉積于近端腎小管上皮導致腎小管損傷的一類疾病,幾乎所有病例與κ輕鏈有關[21]。電鏡下κ輕鏈在近端腎小管上皮形成結晶樣物質沉積為其特征[13]。文獻報道 LCPT占單克隆Ig輕鏈相關性腎臟疾病的4%~5%,低于AL(48.8%)、LCDD(15.2%)[22]。STOKES等[23]報道45例LCPT患者中結晶型40例(87%),有17例(38%)具有范可尼綜合征的一種或多種特征,多數患者有腎功能不全(83%)和/或蛋白尿(98%),伴電解質紊亂或腎小管酸中毒等。目前LCPT的治療主要為硼替佐米基礎方案化療。
3 MGRS的病理機制及診斷
在MGRS的腎臟病理中,存在單克隆Ig組織沉積和無組織沉積兩種病理類型。最常見的是單克隆Ig直接沉積引起的腎損傷。某些MGRS患者腎活檢中無法證實單克隆Ig存在,支持MGRS的間接機制。單克隆Ig可能作為自身抗體誘發機體炎癥或免疫性腎損害。克隆指導的治療可改善這部分患者腎臟預后,使得間接機制得到了進一步的證實[1,13,23-24]。
MGRS是由低級別淋巴PC增殖性疾病分泌的單克隆Ig介導的腎損害,為疾病早期階段,尚不足以診斷為MM或淋巴瘤[1]。MGRS包括AL、LCDD、LCPT、POEMS綜合征(P為進行性多發性神經病;O為臟器腫大,肝脾腫大;E為內分泌病;M為M蛋白;S為皮膚色素沉著)、冷球蛋白血癥和C3腎小球腎炎等[13],其診斷需要腎臟病理專家會診,特別是需借助電鏡或質譜技術確診。WEISS等[18]對2 935例既往診斷為MGUS的患者進行回顧性研究,其中44例(1.5%)目前可診斷為MGRS,分析發現MGRS較MGUS 更易進展為MM(18% 與3%,P<0.001)。在治療管理方面,MGUS與MGRS治療原則明顯不同,MGUS患者一般在3~6個月門診復查M蛋白等,無需特殊治療。而MGRS患者一旦診斷,則需要靶向治療,快速抑制引起腎損害的單克隆Ig的產生,以保護腎功能[1]。
4 IKMG關于MGRS的診斷流程和治療推薦
在臨床上,對蛋白尿或腎病綜合征、腎病合并單克隆Ig、療效不佳的“腎小球腎炎”患者需要考慮MGRS的診斷。2019年國際IKMG發表了關于MGRS的診斷共識,提出對年齡50~70歲存在單克隆M蛋白血癥和無法解釋腎臟疾病的患者,建議進行腎活檢,遵循以下流程[1,25-26]:(1)行腎活檢病理檢測以明確腎損害的模式并顯示單克隆Ig;(2)鑒定血清或尿中相應的單克隆M蛋白;(3)顯示產生單克隆M蛋白的潛在細胞克隆;(4)克隆性疾病的腎外表現[13]。需結合腎外表現及腎活檢與高血壓、糖尿病和風濕免疫系統疾病引起的腎損害進行鑒別診斷。對產生單克隆IgG、IgA或輕鏈的PC克隆(非IgM-MGRS)的患者,使用硼替佐米治療有效;當單克隆IgM是表達CD20的B細胞或淋巴PC時,首選以利妥昔單抗為基礎的治療[26]。MGRS雖然可以發展為MM等腫瘤,但如果得到早期診斷和及時治療,平均OS>10年[1]。
MGRS的提出為MGUS合并腎損害的一組疾病的診斷理清了思路。遵循IKMG的診斷共識,對存在MGUS和無法解釋的腎臟疾病患者進行腎活檢,有利于MGRS的診斷[27]。2020年第一版NCCN指南第一次納入了該病的診治流程,這對血液內科、腎內科和腎臟病理學學科的進步有非凡意義,多學科合作研究、共同管理該病,才能更好地延長MGRS患者的OS。
作者貢獻:魏宇君結合患者臨床資料查閱文獻、進行分析總結并撰寫論文;黃仲夏指導選題和設計、負責文章的質量控制、審校和論文修改,對文章整體負責;申曼、張佳佳、詹曉凱和湯然為本研究提供了臨床資料;李新為論文修改提供了建議。
本文無利益沖突。
