同時累及心臟/皮膚2個部位的單純髓外復發多發性骨髓瘤1例
2021-04-23 20:57:28趙瑩華海應朱文艷
中國典型病例大全 2021年3期
趙瑩 華海應 朱文艷
摘要:多發性骨髓瘤(Multiplemyeloma,MM)是一種惡性克隆性漿細胞疾病,而診斷MM時或治療后合并髓外病變,提示患者預后極差,此部分患者治療經驗也匱乏。本文報告1例多發性骨髓瘤患者,初發時無高危因素,治療反應性好,自體干細胞移植半年后,以全身多處皮膚包塊及心包積液為表現的單純髓外病變復發,且腫瘤細胞出現克隆演變,較為少見,現就其臨床特點,實驗室檢查,診療經過等進行分析,以提高對以單純髓外病變為復發表現的多發性骨髓瘤的認識及治療經驗。
關鍵詞:多發性骨髓瘤;自體造血干細胞移植;髓外病變;復發
【中圖分類號】R246.5 【文獻標識碼】A 【文章編號】1673-9026(2021)03-261-02
1.病例資料
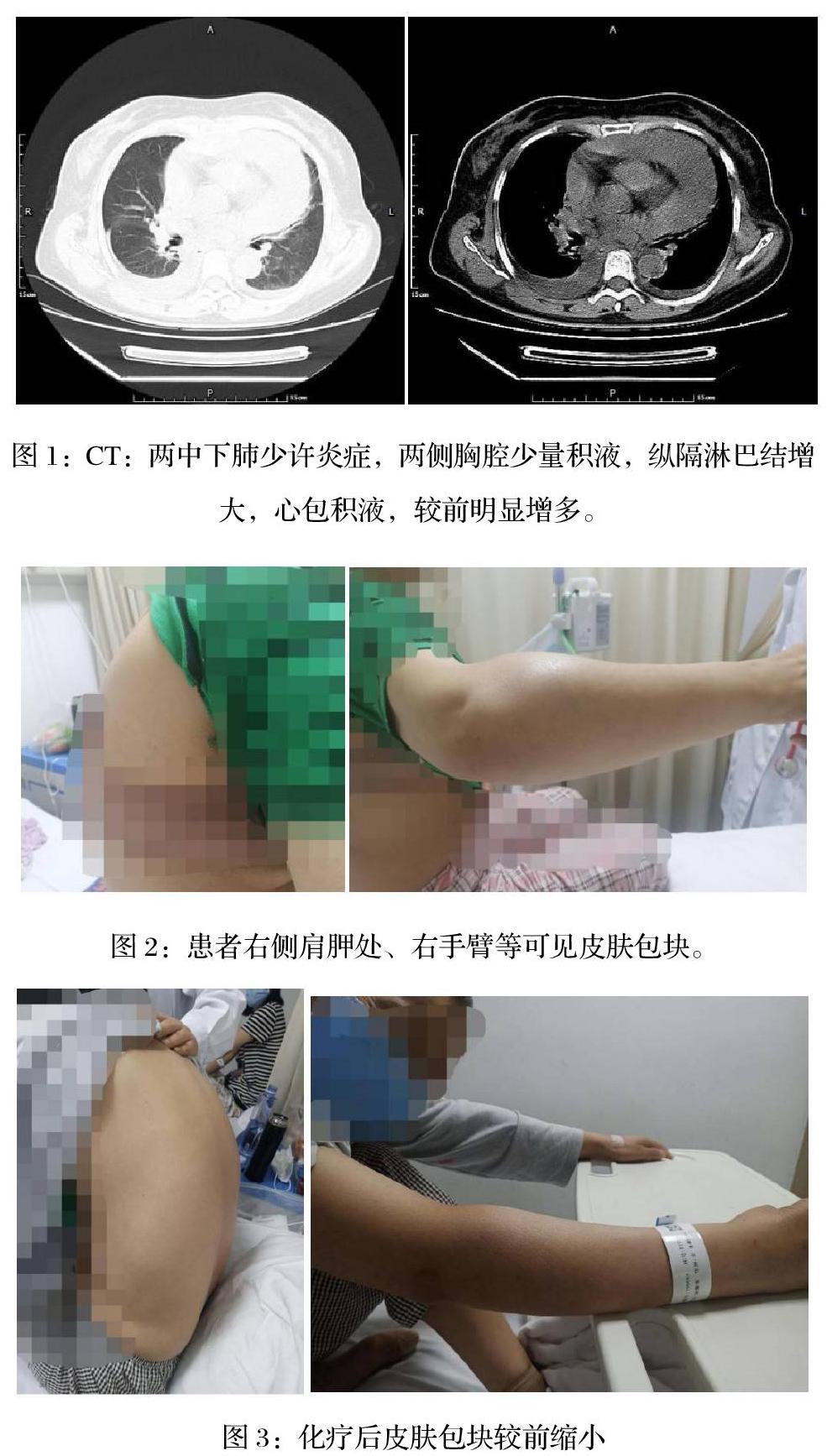
患者柴**,女性,52歲,既往體健。2018年10月因“咳嗽咳痰”就診當地某醫院查肺CT:左肺上葉尖后段不規則形態高密度影(邊緣不整,大小約13*12mm),隨后入住我院胸外科,實驗室檢查:血常規:白細胞 8.6×10^9/L,血紅蛋白:108g/L、血小板223×10^9/L。生化指標:白蛋白 29.60g/L,球蛋白 114.50g/L,尿酸 435.0umol/L,血β2微球蛋白 2.9mg/L,LDH 138U/L。全身CT:1.左側放射冠區小腔隙灶;2.左肺上葉胸膜下結節灶,MT不排除;3.右側第10后肋骨骨質破壞,右側乳腺結節灶,右側肱骨頭,左側肩胛骨,胸腰椎椎體骨質多發穿鑿樣骨質破壞。PET-CT:右側第10肋骨局部骨質破壞,伴異常糖代謝增高,右側肩胛骨下緣,胸骨及左側第10肋骨局部骨質異常糖代謝增高,雙側肱骨頭,脊柱,髂骨,雙側股骨頭密度欠均,伴對稱性糖代謝增高,考慮多發性骨髓瘤?轉移瘤?左肺上葉片狀高密度影,不伴明顯糖代謝增高。血液科會診后考慮多發性骨髓瘤可能,完善相關檢查。診斷:1.多發性骨髓瘤(IgG-κ型)(ISS分期Ⅱ期,DS分期Ⅲ期A組);2.肺結節(性質暫不明確)。2018-11-14、2018-12-12予2次PCD方案化療(硼替佐米 1.3mg/m2,d1,4,8,11;環磷酰胺 0.3g,d1,4,8,11;地塞米松20mg,d1-2,4-5,8-9,11-12),2019-1-14評估為VGPR。2019-1-16、2019-2-21原PCD方案繼續化療,2019-3-20再次評估仍為VGPR(骨穿形態:骨髓增生活躍,成熟漿細胞占0.5%,骨髓MRD:異常漿細胞<0.01%。血免疫固定電泳:在Υ區可見一條單克隆IgG-κ成分。血游離輕鏈:輕鏈κ 9.49mg/L,輕鏈λ 14.8mg/L,κ/λ= 0.6412)。2019-4-2日予環磷酰胺化療后動員干細胞,2019-4-14,2019-4-15采集外周血干細胞,共采集CD34+細胞4.98*10^6/kg。2019-5-14原PCD方案鞏固化療,2019-06-19行胸腔鏡下肺葉部分切除術,術后病理:左肺纖維組織增生及支氣管增生,間質較多平滑肌組織及少量軟骨組織,局部淋巴細胞增生,碳末沉積,考慮錯構瘤伴慢性炎及淋巴細胞增生(排除MM髓外侵犯),術后休養后再次評估本病仍VGPR。2019-7-18、2019-8-15原方案鞏固,7次PCD方案后患者評估達到CR(骨穿形態:MM-CR(骨髓增生活躍,成熟漿細胞占0.5%),MRD:異常漿細胞<0.01% 。血免疫固定電泳:未見異常。2019-10-14予“BUCY”方案預處理[白消安(0.6mg/kg/q6h)41.4mg q6h*d-7至d-4,CTX(1.8g/m2/d)3g d-3至d-2],2019-10-21回輸自體外周血干細胞(MNC 5.57*10^8/kg,CD34+細胞2.48*10^6/kg),回輸后+11天粒系重建,+13天巨核系重建,血象穩定后出院休養,移植后2個月開始口服來那度胺+激素維持治療。2020-4-10入院評估:血常規:WBC 2.9*10^9/L,N 1.4*10^9/L,HB 99g/L,PLT 172*10^9/L,生化指標基本正常。血免疫固定電泳:未見異常。血游離輕鏈:輕鏈κ 63.6mg/L,輕鏈λ 27.5mg/L,κ/λ =2.3127。骨穿形態:MM治療中(骨髓增生活躍低水平,紅系增生活躍,以中晚幼紅細胞增生為主,成熟紅細胞大小不一,成熟漿細胞占1%)。MRD:未見異常漿細胞表達。肺CT:左上肺術后改變,右肺中下葉小斑點,較前(2019-11-05)大致相仿,兩肺少許慢性炎性,心包少量積液。全腹CT平掃+增強:肝臟囊性灶,膽囊壁略增厚,兩腎小結石,左腎腎上腺略增粗,右側腰背部皮下結節灶(長徑約16mm),較前(2019-10-8)為新發,雙側肩胛骨下角,胸腰椎椎體,骨盆骨質多發破壞,符合多發性骨髓瘤表現,較前(2019-10-8)大致相仿,另見心包少量積液。2020年4月底患者右手臂疼痛,局部可觸及包塊,當地醫院查X線提示局部骨質破壞,病理性骨折。隨后患者有胸悶氣急不適,伴惡心嘔吐,2020-5-1于急診對癥處理,2020-5-2收入病房,入院后查體:血壓 105/89mmHg,心率107次/分,心音低。多處皮膚包塊:右側胸部,右側手臂,右側背部肩胛骨處均可觸及皮膚包塊,質地硬,無壓痛,不周圍皮膚組織界限不清,兩肺呼吸音粗,肺底可聞及濕啰音。急診肺CT:左肺術后改變,兩中下肺少許炎癥,兩側胸腔少量積液,較前(2020-4-9)新增炎癥及積液,縱隔淋巴結增大,心包積液,較前明顯增多,右側胸壁皮下多枚小結節灶,較前新增(如下圖1),床邊B超提示:心包積液,心包腔內見液性暗區,左室后壁后方寬22mm,右室前壁前方寬21mm,左室側壁側方約24mm,心尖部約30mm。B超:后背部、右前臂、右胸部、左上臂均可見大小不等皮膚包塊(如圖2)。實驗室檢查:血常規:白細胞:8.0×10^9/L、血紅蛋白:89g/L、血小板:203×10^9/L生化:谷丙轉氨酶:140U/L、谷草轉氨酶:115U/L,乳酸脫氫酶:295U/L,血β2微球蛋白:4.3mg/L;免疫球蛋白:免疫球蛋白G 11.1g/L,免疫球蛋白L輕鏈 4.96g/L,免疫球蛋白κ輕鏈 8.69g/L。心臟標記物:BNP 362pg/ml,余指標正常。心包積液檢查結果:1.常規:外觀血性,比重 1.034,白細胞38000/L,N 90%,L 10%,李凡他實驗陽性;2.生化:總蛋白48.60g/L,乳酸脫氫酶3106U/L,腺苷脫氨酶61U/L;3.找脫離細胞:可見高度可疑惡性細胞;4.腫瘤檢測多項:糖類抗原CA-125:500.7U/ml、神經元特異性烯醇化酶(NSE):45.33ng/mL、細胞角蛋白19片段(CYFRA211):172.2ng/ml,余在正常范圍;5.MM免疫分型:異常漿細胞群占有核細胞81.91%,符合漿細胞腫瘤表型;6.染色體:可見異常克隆性異常-2,del(2q),-10,add(12q),-13,i(17q),-21,+2mar1,+mar2。7.MM-FISH:ATM(11q22.3)、IGH/MAF、IGH/MAFB陰性,TP53(CEP17)陽性。皮膚包塊穿刺病理:符合多發性骨髓瘤累及,免疫組化:CD38(+),CD138(+),CD45(LCA)(散在+),CD20(-),KI-67(95%),CD79a(部分+),Lambda(-),Kappa(+)。骨髓形態:MM-CR(骨髓增生活躍,漿細胞占1.5%),MRD:未見異常漿細胞表達。染色體:未見異常克隆性異常。血免疫固定電泳:未見異常。血游離輕鏈:輕鏈κ 307.5mg/l,輕鏈λ 19.6mg/l,κ/λ 15.6888。診斷多發性骨髓瘤(單純髓外復發伴CEP17缺失)。治療上予心包穿刺引流,對癥支持,2020-5-13予減低劑量的“DECP”方案化療,皮膚包塊縮小(如下圖3),心包積液減少。2020-6-9予“D-PACE”方案化療,化療后患者皮膚包塊繼續縮小,但2周左右再次增大,且增長速度快。2020-7-8日再次入院,出現新發癥狀:右側腹部疼痛,右手臂疼痛。CT提示:兩側胸壁、腹壁皮下多發結節灶,雙腎區及右側結腸旁溝區軟組織影,右心膈角至左心膈上軟組織影,心臟B超未見心包積液。2020-7-9再次予“硼替佐米+D-PACE”化療控制包塊生長,化療過程中患者右側腹痛有緩解,一周后再次出現上述癥狀,后患者及家屬放棄治療出院,2020年8月中旬于家中死亡。
2.討論
多發性骨髓瘤(MM)是一種惡性克隆性漿細胞病,好發于中老年人,目前仍是一種無法治愈的疾病[1]。大部分MM患者表現為骨髓累及的“液體腫瘤”,少數則發生“實體瘤”特征的髓外漿細胞瘤,又被稱為髓外病變(EMD)[2]。隨著檢查手段的不斷進步,EMD的檢出率也逐漸升高,其診治也越來越受到重視。EMD主要包括骨旁漿細胞瘤(EMD-B)和軟組織漿細胞瘤(EMD-S)。EMD在整個病程中都可能出現,新診斷MM患者合并EMD時稱為原發性EMD。復發EMD有兩種形式,一是既往無髓外病灶,復發時出現,本例患者即為此類型,另一種診斷時即有EMD,并以EMD形式復發,這占到原發EMD的一半左右。而EMD-B的發生率高于EMD-S,后者累及部位主要有腎臟(27.3%),淋巴結(17.3%),中樞神經系統(10.1%),肺和氣道(6.5%),消化道(5.8%),胸膜和心臟(5.0%)等。絕大部分EMD(93.5%)僅累及1個部位,而累及2個或以上部位的患者少見,且預后顯著不佳。本例患者在復發時心包積液送檢MM-FISH結果有CEP17陽性,初診時并未發現有相關遺傳性改變,說明腫瘤細胞出現克隆演變,且髓外病變同時累及皮膚及心臟2個部位,較為少見,后期甚至累及腎臟結腸等部位,提示其預后極差,生存期不長。
然而,對于漿細胞如何從骨髓中逃逸形成EMD的機制目前仍不明確,有學者認為缺氧狀態和黏附分子改變可能與之相關。研究表明,使用蛋白酶抑制劑的主流療法可殺死組成大多數腫瘤的漿細胞,但其無法觸及祖細胞,祖細胞增殖并成熟后,即便是病情已得到完全緩解的情況下也會重啟疾病進程。在療效較好的患者中,漿細胞失去分泌免疫球蛋白特征,發生非分泌型逃逸(non-secretoryes-cape),另有完整免疫球蛋白復發時僅以輕鏈形式存在,即lightchainescape。逃逸對應著腫瘤生物學行為轉為高度侵襲性,預示耐藥和短生存期。
目前,對于MM髓外病變復發的治療仍十分棘手。關于EMD的治療,尚無前瞻性研究結果,很難提出有針對性的方案。總的原則是按照高危MM進行治療,尤其是EMD-S。近年來,在MM治療上,已開發了許多具有不同作用機制的新藥,包括新一代的PI、IMiD、單克隆抗體、組蛋白脫乙酰酶抑制劑(histone deacetylase inhibitor,HDACI),以及一些如信號轉導調節劑、紡錘體驅動蛋白抑制劑和核因子-κB(nuclear factor-κB,NF-κB)、促分裂原活化的蛋白激酶(mitogen- activated protein kinase,MAPK)、蛋白激酶 B(protein kinase B,PKB)通路阻斷藥等靶向藥物。對于適合移植患者,可進行自體或異基因造血干細胞移植。但異基因造血干細胞移植后高發EMD也讓研究者對這種治療有所顧慮。
本案例患者為初發時未發現有高危遺傳學因素,多次化療后效果較好,自體干細胞移植后應用來那度胺維持治療。但自體移植后半年左右快速復發,以累及心臟及皮膚的髓外病變為表現,且腫瘤細胞出現克隆演變,為高危患者。針對這類患者建議CAR-T或新藥聯合化療,但其因經濟原因均未嘗試,僅以傳統復發化療方案控制疾病進展,效果較差,患者復發4個月左右死亡,疾病復發發展過程符合目前國內外MM髓外病變的相關文獻報道的臨床特征,但其以累及2個部位,且多處皮膚包塊迅速進展的臨床表現目前報道較為少見,本例報道可積累對本疾病單純髓外復發的認識及臨床經驗。
總之,復發的多發性骨髓瘤,尤其合并髓外病變患者的治療仍然是一個難題,CAR-T免疫治療的逐步大量開展、造血干細胞移植技術的逐步走向成熟,同時針對多發性骨髓瘤治療新藥的不斷涌現,以新藥為基礎的單藥或者多藥聯合治療獲得較好的治療效果,都已經成為復發、難治MM患者治療的主流趨勢。但是,以EMD為表現的復發患者,尤其累及大于1個部位的復發患者,預后極差,生存期短,建議以降低腫瘤負荷及提高患者生活質量,相對延長生命為目的,標準化治療方案的提供還將有賴于今后更多的前瞻性研究。
參考文獻
[1]RolligC,Knop S,Bornhauser M.Multiplemyeloma[J].Lancet,2015,385:2197-2208.
[2]黃曉軍、路瑾、常英軍等.中國多發性骨髓瘤診治指南(2020年修訂),中華內科雜志,2020年5月第59卷第5期.
[3]劉佳慧,安剛,王建祥.CAR-T細胞治療在多發性骨髓瘤中的進展。臨床血液學雜志,2019年32卷7期.
江南大學附屬醫院 214000
