
小球藻蛋白/海藻酸鈉互穿纖維的制備與特性
2021-05-08 08:41:18
食品工業 2021年4期
關鍵詞:結構
1.華東理工大學食品科學與工程系,生物反應器工程國家重點實驗室(上海 200237);2.中國農業大學信息與電氣工程學院(北京 100083)
小球藻(Chlorella)是一種單細胞綠藻,也是首先實現商業化利用的微藻之一[1]。小球藻不僅以膠囊、片劑、口服液等形式作為保健品銷售,同時也被廣泛應用于飲料、面條、餅干之中[2]。小球藻中含有大量優質蛋白質,占小球藻粉的40%~70%,其氨基酸組成超過FDA(食品及藥物管理局)頒布的用于人類食品的蛋白質標準,研究證明小球藻蛋白及其水解物具有良好的抗氧化能力[3-5]。然而關于海藻的科學研究及商業化應用多集中于海藻多糖,對于海藻蛋白質的研究有限[6],限制了小球藻蛋白的應用領域。
互穿網絡結構(IPN)是由2個或2個以上的聚合物網絡組成的聚合物結構,這些聚合物網絡在分子層面上相互交錯穿插,2種聚合物之間并無共價鍵連接[7]。如果聚合物體系中只有一種聚合物交聯形成網絡,所得結構稱為半互穿網絡結構(semi-IPN)[7]。與單一聚合物形成的結構相比,IPN結構及semi-IPN結構使產品在機械強度、熱穩定性及吸水能力等多個方面得到改善[8-10]。海藻酸鈉是一種從褐藻中提取出的海藻多糖,是由β-D-甘露糖醛酸和α-L-古羅糖醛酸組成的線性聚合物,在Ca2+等二價陽離子作用下可迅速形成具有“蛋殼結構”的凝膠[11-13],常用于構建互穿網絡結構。對于IPN結構的研究主要集中在組分添加量對于產品性能的影響,對于組分交聯度的研究較少。
試驗以小球藻蛋白和海藻酸鈉為原料,使用谷氨酰胺轉胺酶(TG酶)和氯化鈣分別交聯兩種原料形成網絡結構,通過濕法紡紗制備具有互穿網絡結構的凝膠纖維,并探究TG酶添加量對于凝膠纖維的結構、物化特性及降解過程的影響。通過傅里葉紅外光譜(FT-IR)及小球藻蛋白交聯度的測定驗證制備凝膠纖維的可行性,進而對凝膠纖維中小球藻蛋白的二級結構、凝膠纖維的吸水能力與持水能力進行評價。探究凝膠纖維在蛋白酶作用下的降解過程,并對小球藻蛋白及其酶解物的釋放過程進行分析。試驗首次將小球藻應用于凝膠纖維制作,可為拓展小球藻蛋白的應用領域提供理論基礎。
1 材料與方法
1.1 材料與試劑
海藻酸鈉(南京都萊生物技術有限公司);破壁小球藻粉(蛋白質含量78%,青島科海生物有限公司);谷氨酰胺轉胺酶(2 100 U/g,酷爾化學科技(北京)有限公司);堿性蛋白酶(200 000 U/g,上海騰騫生物科技有限公司);其他常用化學試劑均為分析純。
1.2 儀器與設備
CP213電子分析天平(奧豪斯儀器(上海)有限公司);HHS-I1-1電熱水浴鍋(上海一恒科學儀器有限公司);SHA-BA恒溫振蕩器(常州朗越儀器制造有限公司);DL-5-B離心機(上海安亭科學儀器廠);STARTER2100 pH計(上海華聯醫療器械有限公司);MOV-212F鼓風干燥箱(上海天呈實驗儀器制造有限公司);UV-2000紫外分光光度計(尤尼克(上海)儀器有限公司);FTIR5700傅里葉紅外光譜儀(尤尼克(上海)儀器有限公司)。
1.3 方法
1.3.1 小球藻蛋白/海藻酸鈉互穿結構凝膠纖維制備
采用Sibaja等[14]的方法并稍作修改,通過濕法紡紗制備小球藻蛋白/海藻酸鈉互穿結構凝膠纖維。將小球藻粉溶于100 mL去離子水中,用考馬斯亮藍法[15]測定其中蛋白質含量,標準曲線為Y=5.66X+0.078(R2=0.999 9),調節其中小球藻蛋白質量濃度至1 g/100 mL。向溶液中加入1 g海藻酸鈉,磁力攪拌30 min使其完全溶解。添加TG酶至其濃度分別為0,1.0,2.5,5.0,7.5和10.0 U/mL,置于50 ℃水浴下反應2 h。反應結束后,置于90 ℃水浴5 min滅酶。冷卻至室溫后,以5 000 r/min離心15 min除去氣泡,所得樣品即為紡絲液。
將紡絲液裝入配備18G針頭的注射器中,擠壓至100 mL質量濃度為10 g/100 mL CaCl2溶液中。2 h后,將凝膠纖維取出,放入去離子水中浸泡1 min后取出,35 ℃下干燥24 h,得到小球藻蛋白/海藻酸鈉互穿結構凝膠纖維。凝膠纖維按照其紡絲液中TG酶濃度分別標記為semi-IPN、IPN1、IPN2.5、IPN5、IPN7.5及IPN10。
1.3.2 傅里葉紅外光譜(FT-IR)
將2 mg凝膠纖維與200 mg溴化鉀充分混合后壓片。采用FT-IR5700傅里葉紅外光譜儀對凝膠纖維的結構進行表征,波數范圍4~4 000 cm-1,分辨率4 cm-1。
1.3.3 二級結構分析
以所得紅外光譜圖為基礎,參考謝孟峽等[16]的方法對凝膠纖維中小球藻蛋白的4種二級結構相對含量進行測定。
1.3.4 小球藻蛋白交聯度的測定
小球藻蛋白交聯度的測定按照Mi等[17]提出的方法。溶液Ⅰ配制:1.05 g檸檬酸,10 mL濃度為1.0 mol/L的NaOH溶液,0.04 g SnCl2·2H2O混合后,用去離子水定容至25 mL。溶液Ⅱ配制:1 g茚三酮溶解于乙二醇單甲醚中,定容至25 mL。將溶液Ⅰ與溶液Ⅱ混合,磁力攪拌45 min,避光儲存。
將1 mL紡絲液溶解于去離子水中,定容至100 mL。將1 mL稀釋后紡絲液與2 mL茚三酮溶液混合,100 ℃水浴20 min。冷卻至室溫后,加入3 mL 50%異丙醇,在570 nm波長下測量吸光度。與茚三酮反應后,樣品中游離氨基含量與其在570 nm波長下吸光度呈正比。樣品中游離氨基含量可代入甘氨酸濃度與其在570 nm下吸光度繪制所得標準曲線進行計算Y=2.805X+0.030(R2=0.999 8)。小球藻蛋白交聯度按式(1)計算。
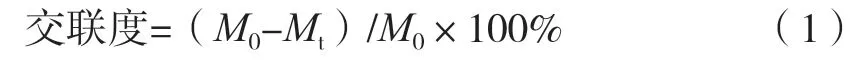
式中:M0為不添加TG酶的樣品中游離氨基物質的量濃度,mmol/L;Mt為不同TG酶添加量的樣品中剩余游離氨基酸物質的量濃度,mmol/L。
1.3.5 吸水能力測定
將一定質量的凝膠纖維置于去離子水中,37 ℃浸泡24 h。取出后用濾紙擦干表面水分,記錄質量。凝膠纖維的吸水性按式(2)計算。
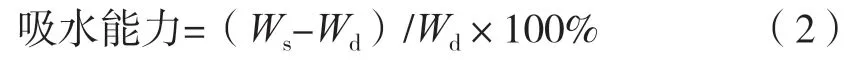
式中:Wd為凝膠纖維干質量,g;Ws為凝膠纖維吸水后質量,g。
1.3.6 持水能力測試
將一定質量的凝膠纖維置于去離子水中,37 ℃浸泡24 h。以5 000 r/min離心10 min,取出纖維,用濾紙擦干表面水分,記錄質量。凝膠纖維的持水性按式(3)計算。
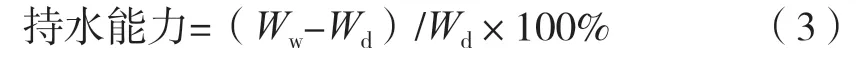
式中:Wd為凝膠纖維干質量,g;Ww為凝膠纖維離心后質量,g。
1.3.7 凝膠纖維的酶促降解
凝膠纖維的酶促降解參照Bidault等[18]的方法并稍作修改。將50 mg凝膠纖維浸泡于100 mL生理鹽水中,調節pH至8.5,加入堿性蛋白酶至400 U/mL。55 ℃水浴下酶解,每隔1 h,吸取2 mL酶解液,于90 ℃水浴5 min滅酶。每次收集樣品后,將2 mL酶溶液加入反應體系來保證酶濃度。試驗結束后,用考馬斯亮藍法[15]測定酶解液中釋放的蛋白質以及蛋白質片段含量。一定時間內小球藻蛋白及其酶解物的釋放率為溶液中釋放蛋白質片段質量與蛋白質初始質量的比值。
1.3.8 數據統計與分析
采用SPSS 19.0分析軟件和Origin 8.0軟件處理所得試驗結果,數據均為3個平行樣本,以平均值±標準差表示,采用ANOVA對數據的差異顯著性進行分析(p<0.05表示差異顯著)。
2 結果與分析
2.1 凝膠纖維紅外測試分析
運用傅里葉紅外光譜從定性的角度分析凝膠纖維制備的可行性,小球藻粉、海藻酸鈉、凝膠纖維的紅外光譜如圖1所示。在海藻酸鈉的紅外光譜中,3 420 cm-1的吸收峰是—OH的伸縮振動;2 929 cm-1的吸收峰是飽和C—H的伸縮振動;1 635與1 417 cm-1的吸收峰則分別對應—COO—的非對稱伸縮振動和對稱伸縮振動[10]。
在小球藻粉的紅外光譜中,酰胺Ⅰ帶(位于1 700~1 600 cm-1代表C==O伸縮振動)與酰胺Ⅱ帶(位于1 590~1 500 cm-1代表N—H彎曲振動與C—N伸縮振動)分別位于1 651與1 554 cm-1處[19]。3 419 cm-1的吸收峰是—OH伸縮振動與N—H伸縮振動重疊的結果[10]。
TG酶添加量不同時凝膠纖維的紅外光譜均表現出海藻酸鈉與小球藻粉的特征吸收峰,說明兩者具有良好的相容性[20]。與semi-IPN相比,具有IPN結構的凝膠纖維在1 554 cm-1的吸收峰強度略有增強,在1 631 cm-1的吸收峰強度明顯增強,說明TG酶成功催化谷氨酰胺與賴氨酸進行共價交聯,生成酰胺鍵[21],形成小球藻蛋白網絡結構。具備IPN結構的凝膠纖維均在1 157 cm-1處呈現吸收峰(代表C—O伸縮振動),而semi-IPN并無此吸收峰,這可能是由于TG酶的催化作用引發小球藻蛋白構象改變[19]。
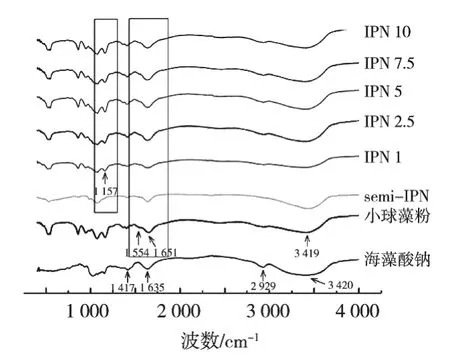
圖1 小球藻粉、海藻酸鈉、凝膠纖維的FT-IR曲線
2.2 小球藻蛋白二級結構分析
通過對小球藻蛋白二級結構進行表征,進一步了解不同TG酶添加量對凝膠纖維結構的影響,結果如表1所示。數據表明,TG酶催化效應對小球藻蛋白二級結構產生顯著影響。與semi-IPN相比,隨著TG酶添加量增加,β-折疊含量從22.83%逐漸下降至17.00%,無規則卷曲含量從15.58%逐漸上升至24.41%。加入TG酶之后,α-螺旋含量總體呈現下降趨勢,TG酶濃度7.5 U/mL時達最低值28.51%。TG酶添加量的改變并未對β-轉角含量造成顯著影響(p>0.05),只是當TG酶濃度1 U/mL時達最高值34.41%,顯著高于其他樣品(p<0.05)。上述現象與Song等[22]研究結果相似。研究顯示,二級結構與蛋白質柔韌性以及強度密切相關。Zhao等[23]研究發現α-螺旋含量下降說明分子內氫鍵含量降低。Liang等[24]研究發現β-折疊的穩定性高于α-螺旋,蛋白質結構的強度很大程度上取決于β-折疊含量。Chen等[25]研究發現無規則卷曲含量增加則代表蛋白質中的高度有序結構展開。結果表明,隨著TG酶添加量增加,凝膠纖維中小球藻蛋白的結構從有序逐漸向無序轉變。
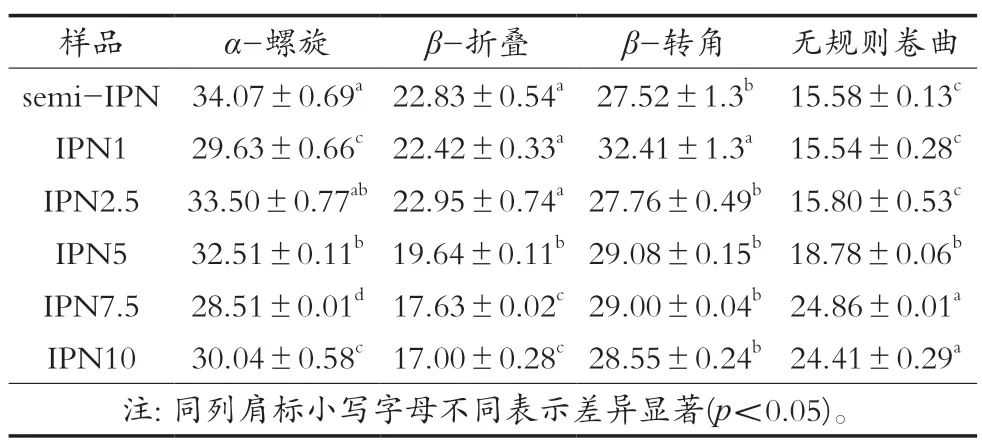
表1 不同TG酶添加量下小球藻蛋白二級結構相對含量 單位:%
2.3 小球藻蛋白交聯度分析
通過測定小球藻蛋白的交聯度,從定量角度分析IPN凝膠纖維制備的可行性。TG酶催化蛋白質之間生成ε-(γ-Glu)Lys共價鍵,從而減少賴氨酸中ε-氨基含量[26]。茚三酮可以測得樣品中游離氨基含量,進而確定交聯度。如圖2所示,隨著TG酶添加量從1 U/mL上升至10 U/mL,小球藻蛋白交聯度從9.67%上升至16.26%,表明TG酶成功催化蛋白質交聯并形成網絡結構,與FT-IR中結果一致。與此同時,海藻酸鈉導致紡絲液黏度增高,阻礙TG酶的催化效應,導致交聯度有限[10]。
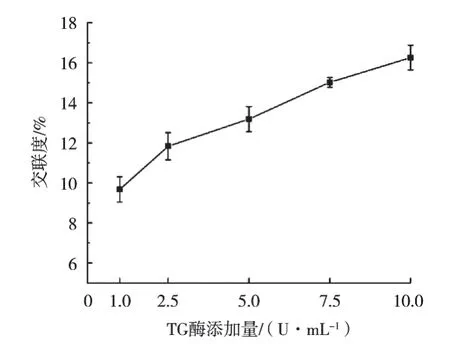
圖2 不同TG酶添加量下凝膠纖維中小球藻蛋白交聯度
2.4 吸水能力分析
吸水能力是衡量材料可否應用于創面敷料的重要參數,不同TG酶添加量的凝膠纖維的吸水能力為82.64%~62.76%,如圖3所示。Shi等[27]認為凝膠纖維的吸水性與其中親水基團的數量及基團親水性強弱密切相關。在凝膠纖維中,水分子與羥基、羧基等親水基團密切結合,表現出較好的吸水能力。隨著TG酶添加量從0上升至10 U/mL,凝膠纖維的吸水性下降24.06%。盡管TG酶的催化作用使小球藻蛋白結構從有序向無序轉變,但是高TG酶添加量使小球藻蛋白的交聯度增加、網絡結構中網格孔徑降低[28]。致密的網絡結構阻止水滲透進入凝膠纖維之中,從而降低吸水能力。
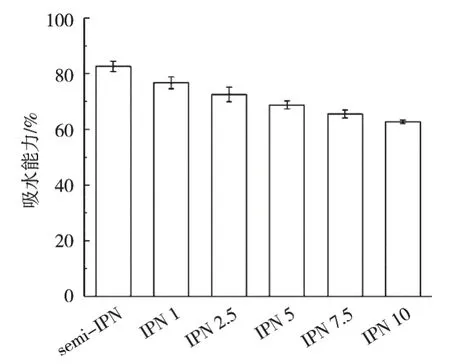
圖3 不同TG酶添加量對凝膠纖維吸水性的影響
2.5 持水能力分析
持水能力是影響吸水材料性能的關鍵因素之一,不同TG酶添加量的凝膠纖維的吸水能力為45.98%~59.48%,如圖4所示。水分析滲透進入凝膠纖維之中,與海藻酸鈉、小球藻蛋白形成穩定的氫鍵,或與親水基團緊密連接,表現出較好的持水能力。隨著TG酶添加量從0上升至10 U/mL,凝膠纖維的吸水能力上升29.36%,這是由凝膠纖維的結構與強度共同導致[29]。TG酶添加量增加催化小球藻蛋白進一步交聯形成更加致密、均勻的網絡結構,可有效防止水分流失。與此同時,TG酶催化形成的ε-(γ-Glu)Lys共價鍵強度是氫鍵和疏水作用力的20余倍[30],因此保證凝膠纖維的持水性。
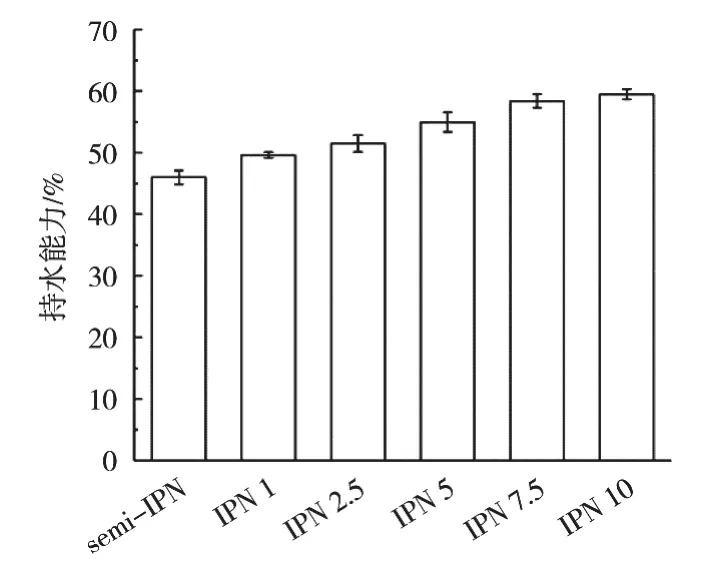
圖4 不同TG酶添加量對凝膠纖維持水能力的影響
2.6 凝膠纖維的酶促降解過程
經過15 h的降解過程,凝膠纖維部分降解,最終形成細小、均勻的碎片,說明海藻酸鈉與小球藻蛋白在凝膠纖維中均勻分布[18]。溶液中的Na+與凝膠纖維中的Ca2+發生置換反應,海藻酸鈉網絡裂解[31],由于不能被堿性蛋白酶催化降解,便以碎片的形式存在。所有樣品中的小球藻蛋白在降解14 h后均完全釋放至酶溶液中,說明盡管小球藻蛋白部分交聯,但仍對堿性蛋白酶敏感,使其可以滲透進海藻酸鈉網絡結構,催化小球藻蛋白網絡結構的裂解[18]。不同TG酶添加量的凝膠纖維在降解過程中的蛋白質組分(小球藻蛋白及其酶解物)釋放率如圖5所示。盡管不影響最終釋放量,但TG酶添加量增加明顯減緩蛋白質組分的釋放速率(除semi-IPN凝膠纖維外)。TG酶添加量從1 U/mL上升至10 U/mL,蛋白質組分完全釋放的時間從11 h增加至14 h,50%蛋白質組分釋放的時間從4 h增加至11 h。這是由于隨著TG酶添加量增加,凝膠纖維吸水能力降低,阻礙小球藻蛋白網絡與堿性蛋白酶活性位點的結合[32]。Semi-IPN中蛋白質組分的釋放速率并不是最快,可能是由于蛋白酶僅作用于小球藻蛋白,與IPN結構凝膠纖維中雙網絡結構同時裂解相比,其網絡結構降解速度較慢。

圖5 不同TG酶添加量的凝膠纖維中小球藻蛋白釋放曲線
3 結論
制備小球藻蛋白/海藻酸鈉凝膠纖維,通過FT-IR及小球藻蛋白交聯度測試證明凝膠纖維具備互穿網絡結構。二級結構分析顯示,TG酶催化使β-折疊含量減少,無規則卷曲含量增加,小球藻蛋白結構從有序向無序轉變。TG酶添加量的增加賦予凝膠纖維更加致密的網絡結構,其吸水能力減弱,持水能力加強。所有的凝膠纖維在蛋白酶以及生理鹽水作用下在14 h之內完全降解。TG酶含量從1 U/mL上升至10 U/mL,小球藻蛋白及其酶解物的釋放速率逐漸下降。試驗結果有助于擴展小球藻蛋白應用領域,為其深層次加工利用提供理論基礎。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
