茜芷膠囊質量標準提高研究
2021-05-12 05:57:00楊博涵朱旭江
中國民族民間醫藥 2021年7期
關鍵詞:方法
楊博涵 朱旭江
1.天津市醫藥科學研究所,天津 300020;2.甘肅省藥品檢驗研究院,甘肅 蘭州 730070
茜芷膠囊是由白芷、茜草、川牛膝、三七組成的復方制劑,其現行質量標準為國家食品量標準提高,在原標準的基礎上,增加了川牛膝中杯莧甾酮的薄層鑒別,并將原標準中用薄層掃描法測定歐前胡素的方法改為高效液相色譜法。結果表明采用高效液相色譜法測定白芷中歐前胡素(C16H14O4)和異歐前胡素的含量,方法操作簡便、結果準確,可作為控制茜芷膠囊的定性和定量指標。白芷始載于《神農本草經》,為傘形科植物白芷, 或杭白芷的干燥根[1],具有散風除濕、通竅止痛、消腫排膿的作用[2],現代藥理實驗表明白芷中的有效成分之一歐前胡素屬于香豆素[3]的一種,具有顯著的止血、抑菌、消炎作用[4]。最新研究證實白芷中呋喃香豆素類化合物具有甲型流感病毒H1N1(Hemagglutinin 1 Neuraminidase 1)和H9N2(Hemagglutinin 2 Neuraminidase 2)的抗病毒活性[5],初步揭示其抗病毒的作用機理。筆者曾嘗試測定茜芷膠囊中阿魏酸、人參皂苷Rg1、人參皂苷Rb1、人參皂苷R1的含量,結果測得的含量均較低,最終本研究采用TLC法對處方中的川牛膝進行定性鑒別,同時采用高效液相色譜法測定白芷中歐前胡素和異歐前胡素的含量,進行方法學考察,從而建立一個操作簡便、專屬性強、重現性好的質量標準,以用于控制該產品的質量。
1 儀器與試藥
Waters Alliance e2695型高效液相色譜儀(Waters 2998 DAD檢測器,Empower色譜工作站,美國Waters公司);CAMAG Scanner-3型薄層掃描儀,CAMAG半自動點樣儀(瑞士CAMAG公司);KH-500DE超聲波清洗器(上海超聲波儀器廠);Mettler AE 240 電子天平(瑞士Mettler公司);R200D型分析天平(德國Sartorius公司); CAMAG Scanner-3型薄層掃描儀。硅膠G薄層板(青島海洋化工廠);中性氧化鋁柱(100~200目)。
乙腈(色譜純,德國Merk 公司)、石油醚(60~90℃)、乙醚、正丁醇、乙酸乙酯、硫酸、乙醇、甲醇為分析純,水為超純水。歐前胡素(批號:110826-201013,含量99.6%)和異歐前胡素(批號:110827-201109,含量99.7%), 均為中國食品藥品檢定研究院提供。茜芷膠囊(批號:J20160901、J20160902、J20160903、J20160707、J20160829),均由甘肅新蘭藥藥業有限責任公司提供。白芷對照藥材(批號:945-9903),三七皂苷R1對照品(批號:110745-200617),茜草對照藥材(批號:121049-201003),川牛膝對照藥材(批號:121065-201105),歐前胡素對照品(批號:110826-201013),杯莧甾酮對照品(批號:111804-201303)。
2 方法與結果
2.1 TLC鑒別 川牛膝[6]TLC鑒別 取本品內容物約2 g,加甲醇50 mL,加熱回流1 h,濾過,濾液濃縮至約1 mL,加于中性氧化鋁柱(100~200目,4 g,內徑為1 cm)上,用甲醇-乙酸乙酯(1∶1)40 ml洗脫,收集洗脫液,蒸干,殘渣加甲醇1 mL使溶解,作為供試品溶液。取杯莧甾酮對照品,加甲醇制成每lml含0.5 mg的溶液,作為對照品溶液。按處方比例及工藝,取不含川牛膝的陰性樣品同供試品溶液的制備方法制成陰性對照溶液。照薄層色譜法(《中國藥典》 2015 年版四部通則 0502)試驗,吸取上述三種溶液各5 μL,分別點樣于同一硅膠G薄層板,以三氯甲烷-甲醇(10∶1)為展開劑展開,取出,晾干,置紫外光燈(365 nm)下或日光下檢視。供試品色譜中,在與對照品色譜相應的位置上,紫外光燈(365 nm)下,顯相同顏色的熒光斑點;日光下,顯相同顏色的熒光斑點,陰性無干擾。結果如圖1所示。
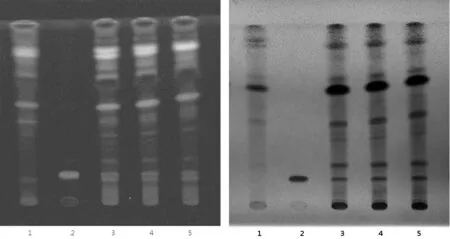
溫度:22℃,相對濕度:45%1. 陰性對照;2. 杯莧甾酮對照品;3. 供試品(批號: J20160707);4. 供試品(批號: J20160901);5. 供試品(批號: J20160902)圖1 川牛膝(杯莧甾酮)薄層鑒別圖譜
2.2 含量測定
2.2.1 色譜條件 色譜柱:資生堂CAPCELL PAK C18( 4.6 mm×250 mm,5 μm);流動相:乙腈-水(50∶50);檢測波長:300 nm;柱溫:30 ℃;流速:1.0 mL·min-1。
2.2.2 對照品溶液的制備 精密稱取歐前胡素對照品11.06 mg,置50 mL容量瓶中, 加甲醇稀釋至刻度,作為對照品儲備液1。精密稱取異歐前胡素對照品13.44 mg,置100 mL容量瓶中, 加甲醇稀釋至刻度,作為對照品儲備液2。再分別精密量取上述兩種對照品儲備液5 mL置100 mL容量瓶中,加甲醇稀釋至刻度,制成混合對照品溶液。
2.2.3 供試品溶液的制備 取本品內容物10粒,混勻,研細,精密稱取約0.5g,置25 mL量瓶中,加入甲醇適量,密塞,超聲處理30 min(功率200 W,頻率60 KHz),放至室溫后,用甲醇稀釋至刻度,搖勻,用0.45 μm微孔濾膜濾過,取續濾液即得。
2.2.4 陰性對照溶液的制備 按處方比例及工藝,制成不含白芷的陰性樣品,取陰性樣品按上述供試品溶液的制備方法制成陰性對照溶液。
2.2.5 方法學考察 專屬性考察:按“2.2.1”項下色譜條件,分別精密吸取對照品溶液、供試品溶液及陰性供試品各20 μL,注入液相色譜儀,測得液相色譜圖,結果如圖2所示。結果表明陰性對測定結果無干擾,且歐前胡素、異歐前胡素色譜分離良好。

1.歐前胡素;2.異歐前胡素;A.對照品;B.供試品;C.陰性對照圖2 茜芷膠囊的HPLC圖譜
線性關系考察:取“2.2.2”項下對照品溶液,按“2.2.1”項的色譜條件,分別進樣2.5、5、10、15、20、25、30、40 μL,測定歐前胡素、異歐前胡素的峰面積,以進樣對照品的質量(μg)為橫坐標(X),峰面積的積分值為縱坐標(Y)進行線性回歸,計算回歸方程,見表1。結果表明,歐前胡素和異歐前胡素在線性范圍內具良好的線性關系。

表1 歐前胡素和異歐前胡素的線性回歸方程
精密度試驗:分別精密吸取歐前胡素和對照品溶液,按上述色譜條件下進樣20 μL,連續重復進樣6次,測定對照品峰面積的積分值,歐前胡素、異歐前胡素的RSD值分別為0.8%和1.3%,結果表明此方法精密度良好。
穩定性試驗:取對照品溶液,分別在0、8、12、16、24 h按“2.2.1”項的色譜條件分別進樣20 μL,以對照品峰面積計算RSD分別為1.1%和1.3%。表明歐前胡素和異歐前胡素在24 h內基本穩定。
重復性試驗:取同一批號供試品(批號J20160901)6份,按“2.2.3”項下方法處理,“2.2.1”項的色譜條件測定含量,歐前胡素和異歐前胡素RSD為1.9%和1.2%,結果表明重復性良好。
加樣回收率:取歐前胡素對照儲備液制成110.2 μg·mL-1的溶液,分別精密吸取1.1 mL、1.3 mL、1.5 mL各3份,置25 mL用量瓶中;取異歐前胡素對照儲備液制成67.0 μg·mL-1的溶液,再分別精密吸取0.6 mL、0.8 mL、1.0 mL各3份,分別置上述相應量瓶中,氮吹儀吹干。精密稱取已知含量的茜芷膠囊(批號:J20160901,約0.25g)各9份,分別置上述量瓶中,依照“2.3”項下的方法測定,計算回收率,結果見表2和表3。
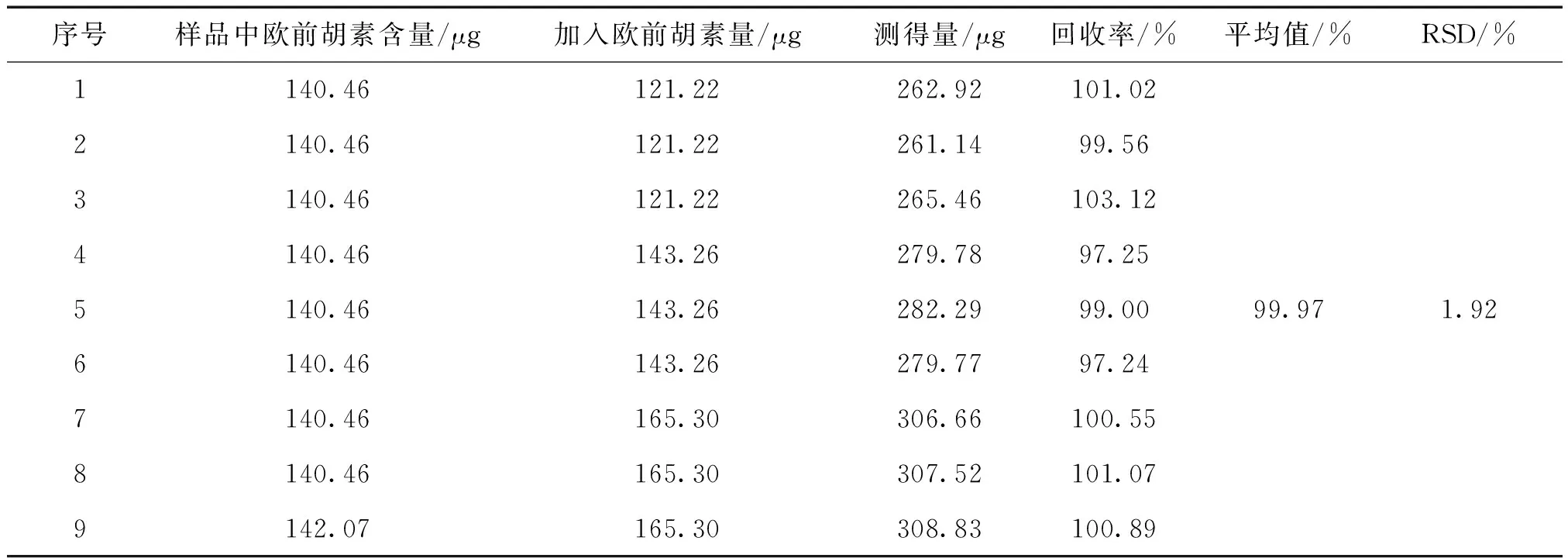
表2 歐前胡素加樣回收率
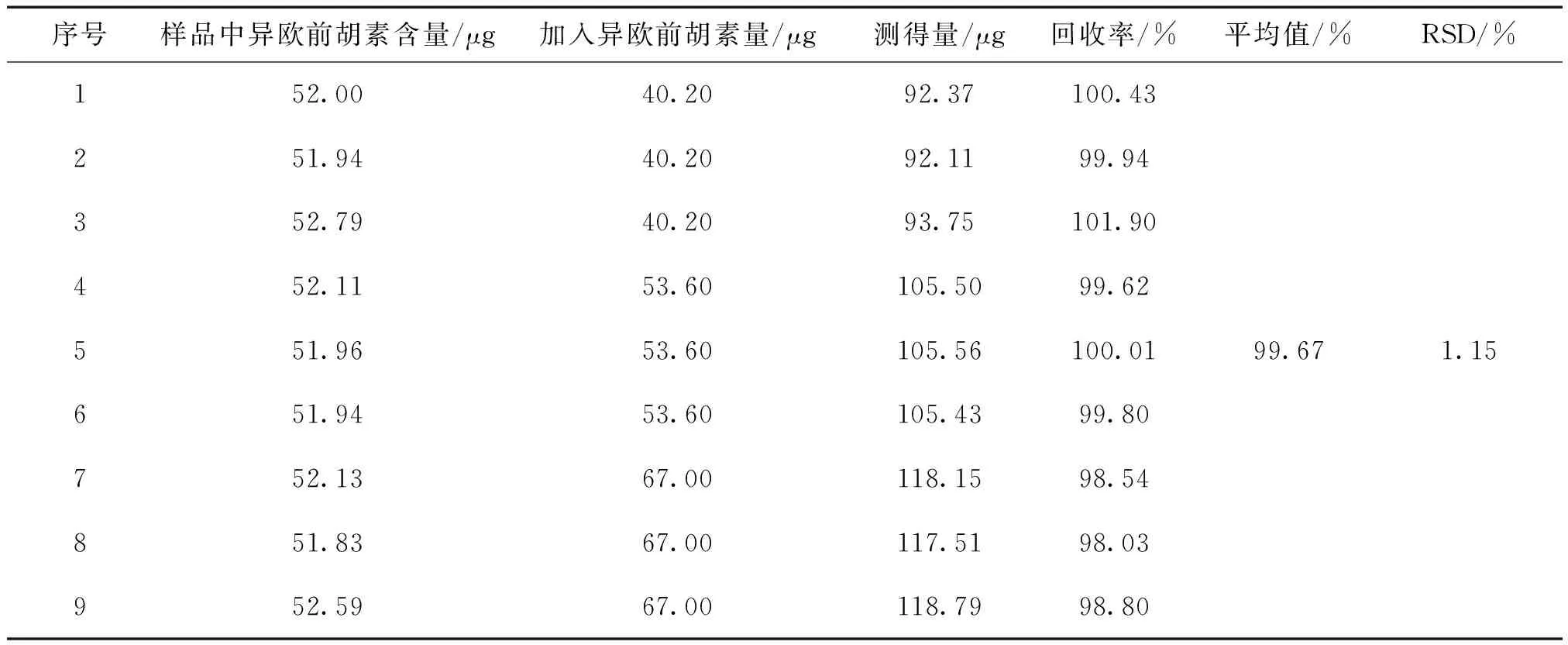
表3 異歐前胡素加樣回收率
2.2.6 樣品測定 按供試品溶液的制備方法和測定方法,對樣品進行制備和測定,分別精密吸取對照品溶液與供試品溶液各20 μL,按“2.2.1”項下色譜條件進樣分析,按外標法以峰面積計算樣品中歐前胡素和異歐前胡素的含量,測定5批供試品的含量,結果見表4。
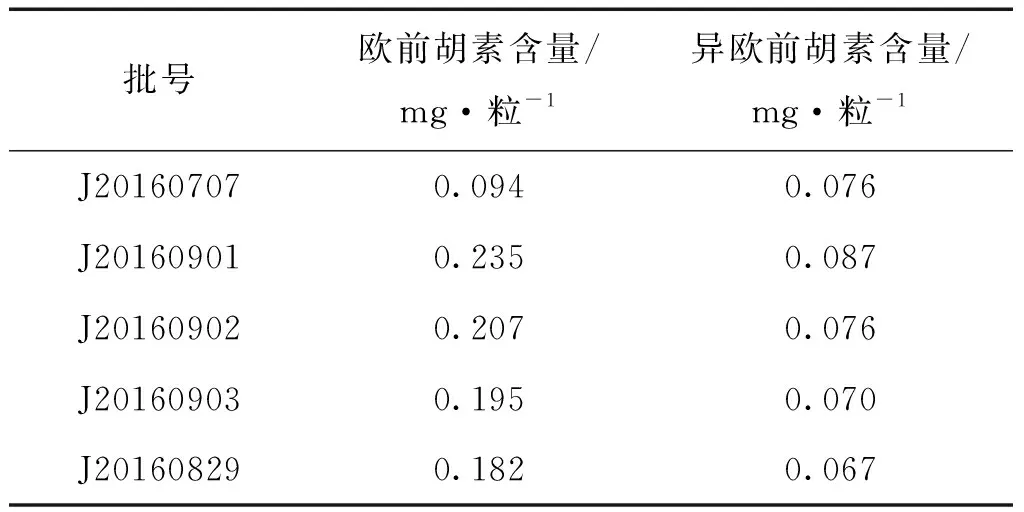
表4 含量測定結果 (n=2)
3 討論
3.1 提取方法的選擇 在樣品的處理階段,提取方法的選擇進行了如下實驗方法進行比較。方法一:按照原標準方法采用石油醚(60~90 ℃)索氏提取,蒸干后用乙酸乙酯定容至5 mL,精密取1 mL,蒸干,再用甲醇溶解后定容至100 mL;方法二:取內容物精密稱取1 g,精密加入甲醇25 mL,稱定重量,超聲30 min,取續濾液20 mL,蒸干,用甲醇溶解后定容至5 mL[6-7];方法三:取內容物精密稱取0.5 g,置25 mL容量瓶中,加入甲醇適量,密塞,超聲處理30 min,放冷,用甲醇稀釋至刻度。將上述三種方法的結果進行比較,發現方法一和方法二在制備的供試品溶液處理上與方法三相比步驟較為復雜,對結果的準確度影響更大。方法三操作更為簡便,無需蒸干步驟,大大縮短實驗所需時間,可以更快速的得出結果。且經過方法三處理后最終結果表明所測成分吸收峰附近無干擾,結果穩定。
3.2 超聲時間選擇 確定樣品提取時間時,按照此方法三步驟進行實驗,在超聲處理時間上分別選取超聲25、30、35、40、50 min,發現超聲30 min即可將茜芷膠囊中白芷的特征成分提取完全,故最終確定30 min為超聲提取時間。
3.3 流動相的選擇 在含量測定時流動相的選擇上,曾先后選用甲醇-水(65∶35)[8-9],乙腈-水(53∶47)[10],這兩種比例為流動相,根據所測成分吸收峰進行調整,比例為乙腈-水(50∶50)時,色譜峰峰形,分離度良好,故選用乙腈-水(50∶50)為流動相。
4 結論
本次質量標準提高實驗,參考中國藥典[11],在原標準的三項鑒別項基礎上增加了川牛膝中杯莧甾酮的薄層鑒別。經過樣品的試驗及方法學驗證研究,結果表明該方法穩定、重現性較好,陰性樣品無干擾。歐前胡素在0.0276~0.4409 μg范圍內呈良好的線性關系,r=0.9999,加樣回收率為99.97%,RSD為1.66%;異歐前胡素在0.0168~0.2680 μg范圍內呈良好的線性關系,r=0.9999,加樣回收率為99.66 %,RSD為1.15%。最終試驗結果表明,該測定方法操作快捷簡便、專屬性強、重復性好,可作為控制茜芷膠囊的定性和定量指標,有效控制本品的質量,實現標準的優化提高。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
