C/g-C3N4/MoS2復合材料的制備及光催化性能
2021-05-16 01:38:52陳建軍李永宇王雅蘋崔天露靳愛玲尚小林
無機化學學報 2021年5期
陳建軍 李永宇 王雅蘋 崔天露 靳愛玲 尚小林 喬 巖
(1鄭州師范學院化學化工學院,鄭州 450044)
(2鄭州輕工業大學材料與化學工程學院,鄭州 450002)
通過C和N原子的sp2雜化,g-C3N4能形成由三嗪環單元組成的高度離域的π共軛體系,從而具有理想的電子結構和穩定性,因此在可見光催化應用方面受到廣泛的關注[1]。然而,由于g-C3N4層間主要是通過弱范德華力相互作用,不利于電子的轉移,從而導致了其光催化效率不高。為此研究人員通過各種方法來改善其光催化活性,如形貌控制[2]、共聚作用[3]、元素摻雜[4]和構建異質結[5]等。最近研究發現碳質材料具有低的逸出功[6],將碳質材料與g-C3N4結合有利于電子和空穴對的分離,進而有望提高光催化性能。為此研究者將g-C3N4與碳納米管(CNT)[7]、富勒烯[8]、石墨烯[9]、碳點[10]和無定形碳[11]等碳質材料進行了耦合,并對其催化性能進行了探究,結果證實兩者的結合確實可以改善g-C3N4光催化活性。
此外,研究發現MoS2結構中的S—Mo—S基團會在邊緣產生不飽和的Mo和S原子,而它們將作為活性位點應用在光催化反應中,有助于光催化效率的提高,在取代貴金屬Pt做助催化劑方面具有巨大的應用潛力。Zhao等[12]和Hou等[13]合成了g-C3N4/MoS2和g-C3N4/石墨烯/MoS2復合材料,并分別研究了它們的光催化活性。到目前為止,大多數研究僅限于MoS2納米片作為助催化劑。與單層MoS2相比,MoS2量子點中有更多的不飽和末端硫原子作為反應活性位點。因此,MoS2量子點作為光催化反應中的輔助催化劑,具有更大的應用潛力。
基于以上分析,本工作將結合碳質材料和MoS2量子點的優勢,進一步改善g-C3N4的光催化性能。首先用一步熱縮合的方法制備了無定形C/g-C3N4,然后采用水熱方法合成了三元的C/g-C3N4/MoS2復合材料,并對MoS2的含量進行了調控,同時研究了三元復合材料在可見光照射下的光催化性能,并對其催化機理進行了探究。
1 實驗部分
1.1 試劑與儀器
尿素、葡萄糖、四硫代鉬酸銨、無水乙醇和N,N-二甲基甲酰胺均購于國藥集團化學試劑有限公司,所用試劑均為分析純。蒸餾水為自制。
采用傅里葉變換紅外光譜分析儀(FTIR,FTIR-650)分析樣品中存在的官能團。采用X射線衍射儀(XRD,Rigaku Uitima Ⅳ)分析樣品的物相組成(CuKα,λ=0.154 06 nm,U=40 kV,I=40 mA,2θ=10°~80°)。采用透射電子顯微鏡(TEM,JEOL 2100)對樣品的形貌進行表征(工作電壓為200 kV);采用比表面分析儀(BET,Autosorb iQ)對樣品的低溫氮吸附-脫附性質進行表征。采用紫外可見分光光度儀(UVVis,Cary-5000)分析樣品的光吸收性能,其中參比物為固體硫酸鋇。采用熒光光譜儀(PL,F-4600)測定樣品的熒光光譜。采用X射線光電子能譜(XPS,ESCALAB-150,AlKα靶)檢測樣品的化學組成和元素價態。利用電化學工作站(CHI660E)測試樣品的電化學性質。
1.2 光催化劑的制備
1.2.1 g-C3N4的制備
稱取一定量的尿素置于陶瓷坩堝,置于馬弗爐中500℃下煅燒2 h,冷至室溫得到g-C3N4,所得樣品記為CN。
1.2.2 C/g-C3N4的制備
將10 g尿素和0.005 0 g葡萄糖均勻混合后置于陶瓷坩堝,500℃下保溫2 h,冷卻至室溫,獲得的樣品標記為CCN。
1.2.3 C/g-C3N4/MoS2的制備
將一定量的(NH4)2MoS4和60 mg的CCN溶解于60 m L DMF中,超聲40 min后將該溶液轉移至100 mL反應釜,在200℃下反應15 h。待反應釜冷卻至室溫后,離心分離并用去離子水洗滌,60℃下干燥即得到C/g-C3N4/MoS2(CCN/MoS2)。按上述方法依次制取MoS2質量分數為1.5%、2.0%、2.5%的CCN/MoS2復合催化劑,所得樣品分別命名為CCN/MoS2-1.5%、CCN/MoS2-2.0%、CCN/MoS2-2.5%。材料制備流程如圖1所示。
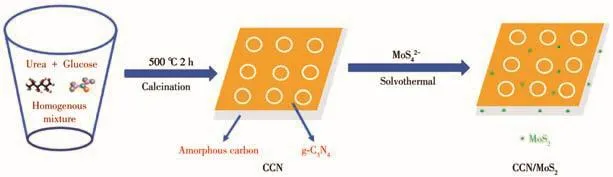
圖1 CCN/MoS2的制備流程圖Fig.1 Illustration for synthesis of CCN/MoS2
1.2.4 CN/MoS2的制備
除了用CN來替代CCN以外,CN/MoS2的制備流程和CCN/MoS2相同(MoS2質量分數為2.0%),所得樣品記為CN/MoS2-2.0%。
1.3 光電化學測試
使用300 W氙燈作為光源,以Pt電極為對電極,Ag/AgCl為參比電極,0.1 mol·L-1Na2SO4溶液為電解質形成三電極體系,對樣品進行光電流測試。
1.4 可見光下光催化降解性能測試
將100 mg催化劑加入到100 mL 10 mg·L-1甲基橙溶液中,暗反應0.5 h后,取第一個樣。開冷凝水,開氙燈(采用420 nm的濾光片獲取可見光),然后每隔30 min取樣一次,共取4次。每次取樣體積均為10 mL。取樣后將樣品置于避光處。最后將取得的5個樣品離心分離15 min,吸取上層清液測其吸光度,根據吸光度變化計算催化劑對甲基橙的降解率。
2 結果與討論
2.1 XRD分析
圖2是制備的樣品的XRD圖。從圖中可以看出,所有樣品在13.3°和27.4°處都有2個明顯的衍射峰,其中13.3°處的衍射峰為三嗪相結構形成的(100)面,27.4°處為芳香體系中共軛雙鍵堆疊形成的(002)面,證明所有樣品中都有石墨相氮化碳(g-C3N4)晶體結構的存在。與CN相比,CCN的衍射峰位置并沒有發生明顯的移動,表明C不是以摻雜形式進入到g-C3N4晶格中[14],而是兩者形成了CCN復合物[11]。在CCN中并沒有觀察到無定形碳峰的存在,可能是因為無定形碳含量太低的緣故。另外,由于復合物中的MoS2含量太少,CCN/MoS2樣品中沒有觀察到MoS2的物相特征峰。
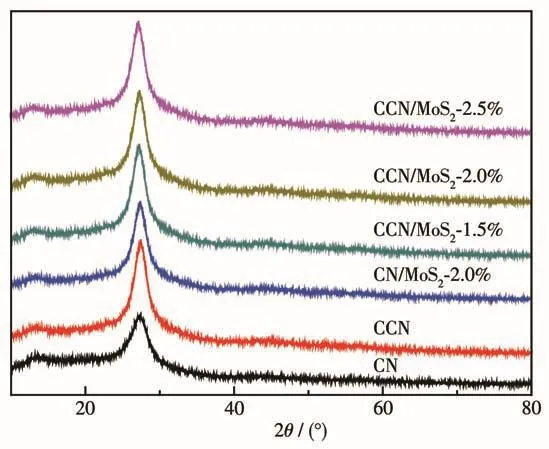
圖2 制備的樣品的XRD圖Fig.2 XRD patterns of as-prepared samples
2.2 FT-IR分析
圖3為制備的樣品的紅外光譜圖,其中CN在809 cm-1處的吸收帶代表三嗪結構的振動特性。1 634、1 564和1 420 cm-1處強烈的特征峰,可歸因于芳香結構中C—N的骨架振動[15]。1 249和1 327 cm-1處的吸收峰,分別對應七嗪雜環單元的N—(C)3和C—N—H的伸縮振動特性[16]。位于3 200~3 400 cm-1處的寬帶是未縮合的氨基與其表面吸附的H2O分子形成的N—H鍵和O—H鍵造成的[17]。CN、CCN和CCN/MoS2具有相似的吸收帶,表明無定形碳和MoS2的引入并沒有改變CN的結構。由于MoS2含量太低,紅外譜圖中也沒有出現MoS2的特征峰。
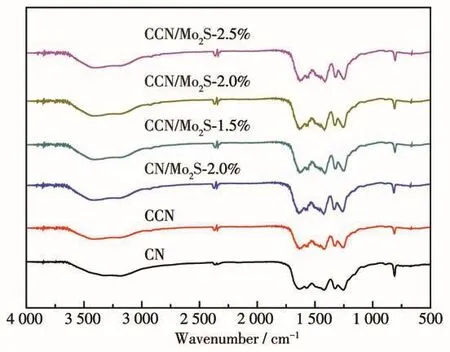
圖3 制備的樣品的紅外譜圖Fig.3 FTIR spectra of as-prepared samples
2.3 XPS分析
為了進一步分析樣品的元素組成和存在狀態,對樣品進行了XPS的表征,結果如圖4和表1所示。從圖4a中可以看到,C1s譜圖在288.1、286.2和284.8 eV處具有3個不同的峰,分別對應于芳香結構中(N—C=N)sp2雜化的碳、殘留的C—O和石墨C—C鍵。另外從圖中可以看出,與CN相比,CCN/MoS2-2.0%中C—C和C—O鍵的相對強度增強,而N—C=N的相對強度則減弱,表明無定形碳與CN存在一定的相互作用。樣品的高分辨N1s譜圖(圖4b)在398.5、399.6、401.2和404.3 eV處的峰,分別對應于sp2雜化的N(N—C=N)、sp3雜化的N(N—(C)3)、端氨基中N(C—N—H)和七嗪環的電荷效應[18]。從表1中可以看出,與CN中的碳氮原子比(nC/nN=0.75)相比,CCN/MoS2-2.0%中nC/nN(1.04)有了明顯提高,進一步證明了無定形碳和CN實現了結合。圖4c為Mo3d的高分辨譜圖,在231.1和232.3 eV處的峰分別對應于Mo3d5/2和Mo3d3/2,根據文獻報道[19],這2個峰是MoS2中Mo4+的特征峰。在235.4 eV位置出現的峰則對應于Mo6+,可能是因為部分Mo4+在表面的氧化引起的。圖4d是S2p的高分辨譜圖,在160.1和163.3 eV處的峰對應于S2p3/2和S2p1/2,證明了S2-的存在,而在168.3 eV出現的峰是部分硫氧化的結果造成的[20]。結合以上分析可知成功地制備了CCN/MoS2復合材料。
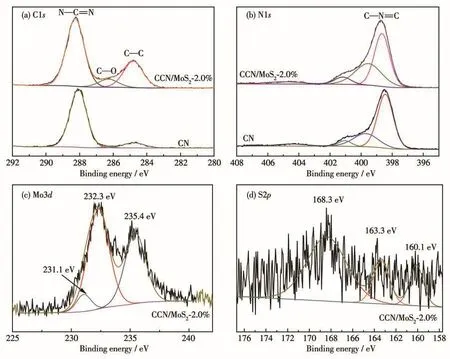
圖4 CN和CCN/MoS2-2.0%的XPS譜圖Fig.4 XPS spectra of CN and CCN/MoS2-2.0%
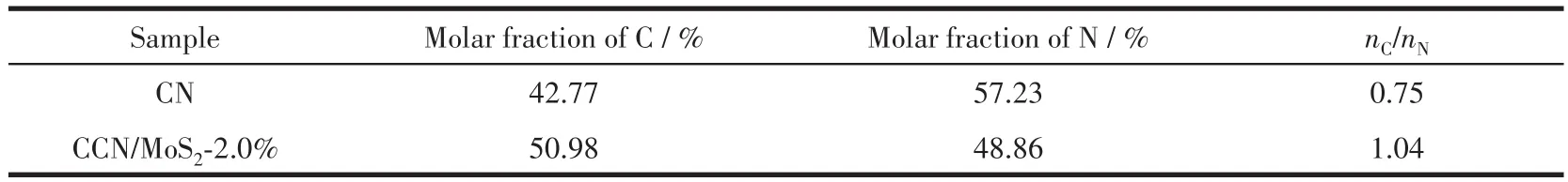
表1 CN和CCN/MoS2-2.0%中的n C/n NTable 1 n C/n N of CN and CCN/MoS2-2.0%
2.4 微觀結構的分析
圖5為CCN/MoS2-2.0%的TEM圖,從圖5a中可以看出,樣品具有重疊層,上面是具有多孔結構的g-C3N4納米片,孔的尺寸為20 nm左右,這種多孔是尿素在加熱過程中產生的氣體擴散形成的,沒有孔的片狀部分則是無定形碳,兩者的界面緊密地結合在一起。這種界面的結合有利于電子的快速轉移,進而減少電子和空穴的復合。圖5b展示MoS2量子點沉積在CCN的表面上,量子點尺寸為2 nm左右。此外,元素的分布圖如圖5c~5f所示,從圖中可以看出,C、N、Mo和S均勻分布在CCN/MoS2-2.0%中,進一步證明了MoS2的存在。

圖5 CCN/MoS2-2.0%的TEM圖 (a、b)和C、N、Mo和S的元素分布圖 (c~f)Fig.5 TEM images of CCN/MoS2-2.0%(a,b)and the corresponding element mapping for C,N,Mo,and S(c~f)
2.5 分級孔結構表征
通過氮氣吸附-脫附曲線來評價樣品的孔結構,結果如圖6所示。從圖6a中可以看出,CCN和CCN/MoS2-2.0%都是典型的Ⅳ型等溫線,具有一個H3型的滯后環,表明樣品中介孔的存在,CCN和CCN/MoS2-2.0%的孔徑分布中心在20 nm左右(圖6b),與TEM分析的結果相一致。通過計算可以得到樣品的比表面積和孔體積(表2),與CCN相比,CCN/MoS2-2.0%具有更大的比表面積和孔體積,能提供更多的反應活性中心,進而有利于光催化反應的進行。
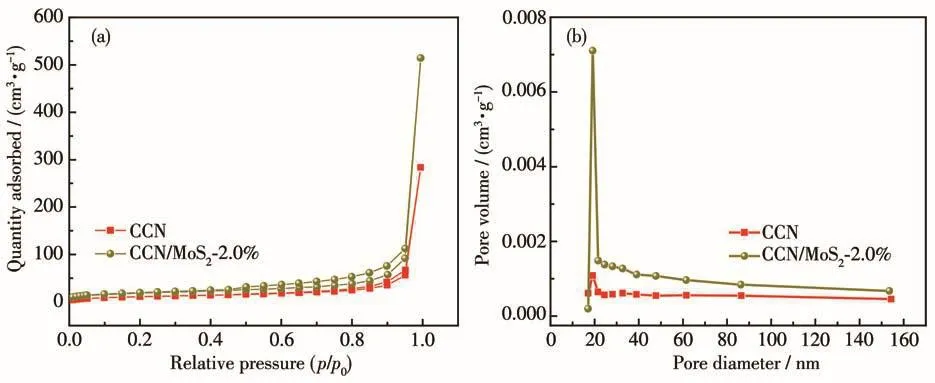
圖6 CCN和CCN-MoS2-2.0%的吸附-脫附等溫線(a)和孔徑分布圖(b)Fig.6 N2 adsorption-desorption isotherms(a)and pore size distribution curves(b)of CCN and CCN-MoS2-2.0%

表2 CCN和CCN/MoS2-2.0%的比表面積、孔徑和孔體積Table 2 Specific surface areas,pore sizes and pore volumes of CCN and CCN/MoS2-2.0%
2.6 光吸收性能分析
光吸收性能是影響材料光催化性能的重要指標之一,樣品的UV-Vis DRS光譜(UV-Vis漫反射光譜)如圖7所示。從圖中可以看出,所有樣品在可見光區都有寬的吸收。與CN、CCN和CN/MoS2-2.0%相比,CCN/MoS2在400~800 nm范圍的光吸收強度出現明顯的增強,并且隨著MoS2含量的增加而增強,這是因為黑色的MoS2對光有較強的吸收造成的。
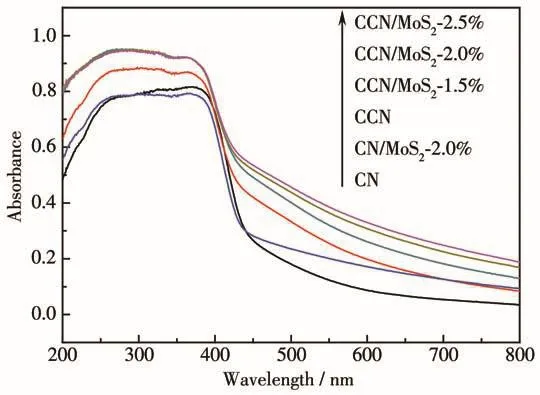
圖7 制備的樣品的UV-Vis DRS譜圖Fig.7 UV-Vis DRS spectra of as-prepared samples
2.7 催化劑的光電性質分析
電子-空穴的分離是影響光催化性能的另外一個重要因素,可通過熒光發射光譜來分析光生載流子的分離能力。圖8是在380 nm波長光激發下,CN、CCN與CCN/MoS2-2.0%熒光發射光譜的結果。從圖中可以看出,相較于CN和CCN,CCN/MoS2-2.0%的熒光強度有了明顯的減弱,說明MoS2作為助催化劑,具有高效捕獲電子的能力,能夠抑制電子和空穴的復合,提高光生電荷之間的分離效率。
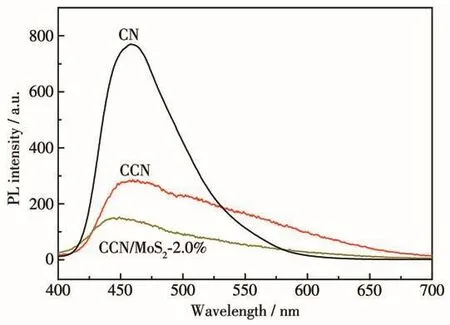
圖8 CN、CCN和CCN/MoS2-2.0%的PL發射譜圖Fig.8 PL emission spectra of CN,CCN and CCN/MoS2-2.0%
通過光電流測試進一步確定MoS2在轉移光生電子中的作用,結果如圖9所示。從圖中可以看出,與CN和CCN相比,CCN/MoS2-2.0%光電流強度得到了明顯的增強,光電流強度的增加,意味著復合后的產物能更有效地分離界面處的光生載流子,進而產生更多的反應活性位點。
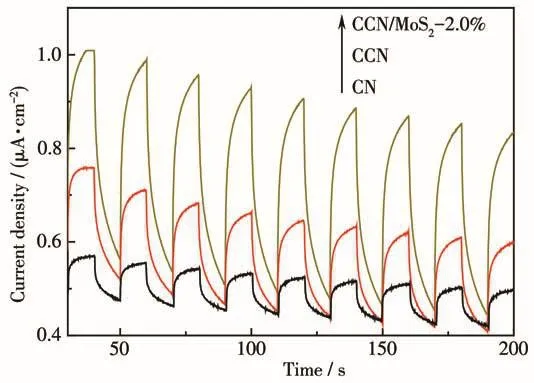
圖9 可見光照射下CN、CCN和CCN/MoS2-2.0%的光電響應圖Fig.9 Transient photocurrent responses of CN,CCN and CCN/MoS2-2.0% under visible light irradiation
2.8 光催化性能分析
選用甲基橙作為目標降解物來評價催化劑的性能。圖10a為可見光照射120 min后制備的樣品對甲基橙的降解率。從圖中可以看出:與CN、CCN和CN/MoS2-2.0%相比,CCN/MoS2對甲基橙的降解率有了明顯的增強,其中,CCN/MoS2-2.0%呈現最優的催化性能,對甲基橙的降解率為65%。當染料的初始濃度非常低時,光催化降解滿足Langmuir-Hinshelwood一級動力學模型[21]:ln(c0/c)=kt,其中c0是光催化降解開始時染料的濃度,k是表觀速率常數,c為經過時間t照射后染料的濃度。按照此模型進行的線性模擬如圖10b所示,從圖中可以看出,CCN/MoS2-2.0%的模擬直線斜率最大,表觀速率常數為0.008 6 min-1,為 CN/MoS2-2.0%(0.001 5 min-1)、CN(0.003 3 min-1)和 CCN(0.003 8 min-1)的5.7倍、2.6倍和2.3倍。綜合以上分析可知,CCN/MoS2-2.0%在同等條件下呈現最優的降解率和最大的反應速率,展示了更高的光催化活性。這可能是以下原因造成的:首先,CCN/MoS2-2.0%具有更強的可見光吸收能力;其次,其具有更高的比表面積,能為催化反應提供更多的反應活性位點;再次,復合后的材料有利于電子和空穴的分離,產生更多的氧化活性中心,從而能夠高效地降解有機污染物。
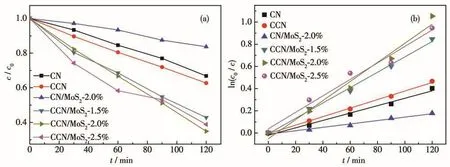
圖10 制備的催化劑在可見光下對甲基橙的光降解曲線(a)及相應的動力學線性模擬(b)Fig.10 Degradation curves(a)of methyl orange by as-prepared catalysts under visible light and corresponding kinetics linear fittings(b)
復合材料可見光下光催化機理如圖11所示。在可見光的照射下,g-C3N4中的電子和空穴發生分離,導帶上的電子首先轉移到活性炭,然后轉移到MoS2表面。MoS2表面上生成的活性集團和g-C3N4上的空穴都具有強的氧化性,能夠高效地降解有機污染物。在整個反應過程中,MoS2作為電子接受體,能夠進一步促進電子轉移,進而有效地抑制電荷的重組。另外從圖10中可以看出,當MoS2含量過高的時候,其催化性能反而降低了,這是因為過多的MoS2反而為電子-空穴復合提供了場所,導致光催化效率降低。
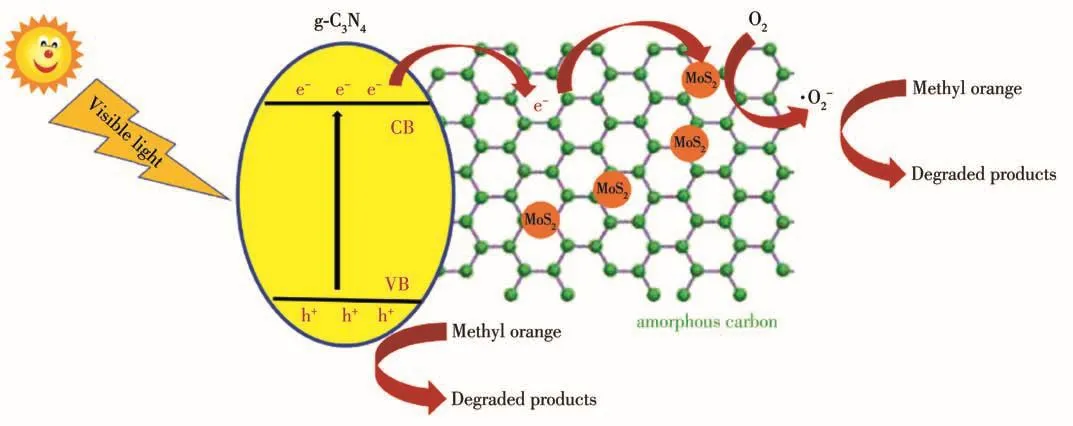
圖11 光催化反應中CCN/MoS2上的光生電荷分離過程Fig.11 Photogenerated charge-separation process on CCN/MoS2 for photocatalytic reaction
穩定性也是評價催化劑性能的重要因素,因此通過循環降解實驗對CCN/MoS2-2.0%的穩定性進行了測試,結果如圖12所示,催化劑重復使用3次后,甲基橙的降解率基本沒有明顯的變化,說明CCN/MoS2-2.0%具有較好的穩定性。
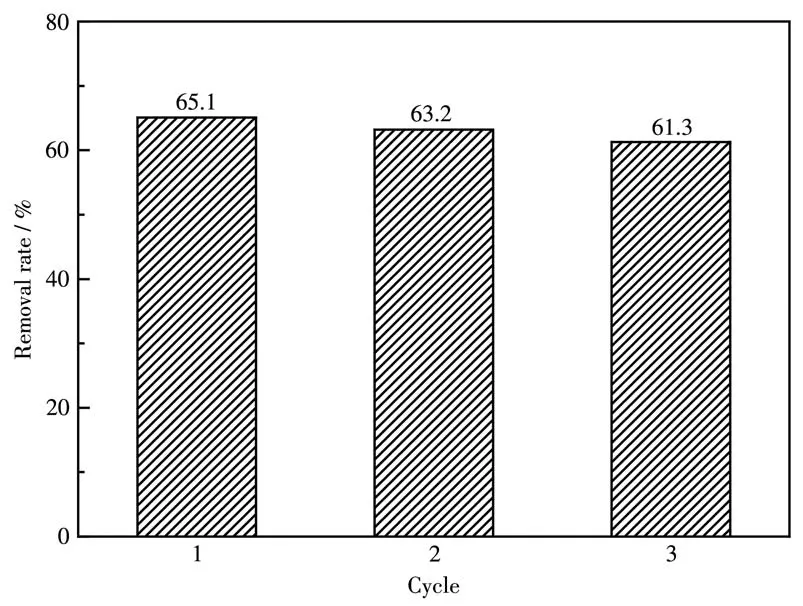
圖12 CCN/MoS2-2.0%可見光降解甲基橙的循環實驗Fig.12 Recycling test of CCN/MoS2-2.0% for degrading methyl orange under visible light
3 結 論
采用熱縮合結合溶劑熱的方法合成了C/g-C3N4/MoS2(CCN/MoS2)三元復合材料。與CCN和CN/MoS2-2%相比,CCN/MoS2-2%具有最優的可見光催化性能,對甲基橙的降解速率常數是CCN的2.3倍,是CN/MoS2-2%的5.7倍。無定形碳的引入不僅能增強CN的可見光吸收性能,提高其比表面積,還有利于促進光生載流子的有效分離。MoS2作為助催化劑,具有高度捕獲電子的能力,能進一步提高光生電子-空穴的分離效率。本工作為合成高性能、易制備的可見光催化復合材料提供了一種有效的途徑。
猜你喜歡
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
電子制作(2018年18期)2018-11-14 01:48:24
纖維復合材料(2018年3期)2018-04-25 07:22:58
電子測試(2017年11期)2017-12-15 08:57:13
山東工業技術(2016年15期)2016-12-01 05:31:22
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
應用化工(2014年10期)2014-08-16 13:11:29

- 無機化學學報的其它文章
- Synthesis and Characterization of Metal-Organic Framework Based on 2,6-Bis(4-carboxybenzylidene)cyclohexanone
- Two Metal-Organic Frameworks Built from 2,2′-Dimethyl-4,4′-biphenyldicarboxylic Acid
- Three Photochromic Co-crystals Based on Viologen Moiety
- Effect of Mass Ratio of Ni and Co in Initial Solution on Oxygen Evolution Reaction Performance of Ni-Co-S-O/NF Catalyst in Alkaline Water Electrolysis
- Structure and Fluorescence Properties of Three 1D/2D/3D Zn/Cocomplexes Based on Flexible Tetracarboxylic Acid
- Two Nitronyl Nitroxide Biradical-Bridged Lanthanide One-Dimensional Chains:Crystal Structure,Magnetic Properties and Luminescent Behavior