
過渡金屬催化CO2/H2參與的羰基化研究進展
2021-06-02 11:39:20華凱敏劉曉放魏百銀張書南王慧孫予罕
物理化學學報 2021年5期
關鍵詞:體系
華凱敏,劉曉放,魏百銀,3,張書南,王慧,*,孫予罕,3,4,*
1中國科學院上海高等研究院,中科院低碳轉化科學與工程重點實驗室,上海 201203
2中國科學院大學,北京 100049
3上海科技大學物質科學與技術學院,上海 201203
4上海低碳技術創新功能型平臺,上海 201620
1 引言
目前,大氣中二氧化碳(CO2)的濃度已達到創紀錄的水平,且在持續增長,這引發了全球氣候變暖、海平面上升和海洋酸化等重大環境問題1。因此減少CO2排放成為國際社會的共識和高度關注的熱點,也是國內產業升級和環境保護的重要指標2,3。另一方面,溫室氣體CO2也是一類理想的C1資源,具有儲量豐富、可再生、廉價易得、無毒等優點。基于此,開發CO2利用技術以獲得高附加值化學品4和燃料5等越來越受到關注,這既減少了碳排放,又促進了經濟的可持續發展。
在CO2利用中,雖然CO2容易與強親核試劑反應,它仍通常被認為是一種“惰性”分子,這是因為CO2已經處于碳的最高氧化態,本身化學性質穩定,其標準吉布斯自由能為ΔG = -394.38 kJ·mol-1,具有熱力學穩定性和動力學惰性的特點。為解決CO2活化動力學上的挑戰,近幾十年來的基礎研究持續關注于催化劑的開發,并取得了顯著進步6-8。在過去的幾十年里CO2利用被廣泛地探索,將CO2轉化為高附加值的化學品和能源產品取得長足的發展6,9。如圖1所示6,根據CO2分子的還原水平和鍵形成過程(C=O鍵被C―H或其他C―元素鍵取代),CO2轉化可分成三類。圖1左側CO2通過非還原性轉化,生成諸如碳酸酯或聚碳酸酯等產品;右側則是CO2被完全還原,生成甲烷(CH4)或飽和碳氫化合物。而中間一類轉化結合了CO2還原和化學鍵形成,可獲得一大類功能化分子,如甲酸、甲醇、醇、羧酸、酰胺、醛等大宗或精細化學品。這一類還原功能化無疑是最豐富多彩同時也最具價值的CO2轉化之一。
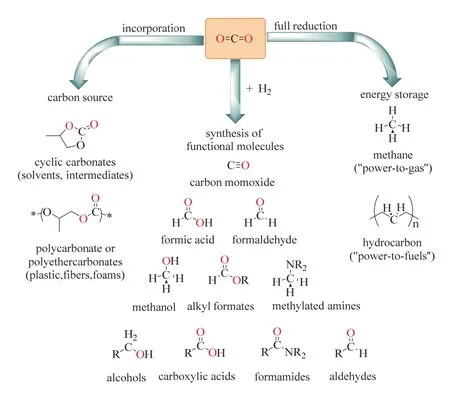
圖1 CO2作C1源的資源化利用Fig. 1 Selective transformations of CO2 as C1 building-block.
為實現CO2的還原轉化,通常需要通過熱還原、光還原或電還原等過程提供較高的能量。CO2的光/電化學還原領域的相關工作也被廣泛報道10,11。在CO2熱催化的化學還原中常見的還原劑有H2、氫硅烷、氫硼烷等12。其中,可以從非化石資源中產生,如通過清潔能源光分解水生成的H2是最具應用前景的還原劑。CO2加氫常見產物有一氧化碳、甲酸、甲烷、甲醇、高級醇、烯烴、液態燃料等4,6,13-21,這些生成的功能產品實現了CO2/H2融入化學品“價值鏈”(圖1,中間路徑)中。其中,CO2催化加氫可以逆水煤氣變換直接得到CO,也可以催化加氫得到CO替代品,如甲酸、甲醛等,這些CO替代品可以進一步分解得到CO。而CO是現代化工生產中不可或缺的大宗化學品,如可參與到羰基化反應中22。羰基化反應是指通過催化的方法,在有機化合物分子內,引入羰基及其衍生基團的一類重要反應。經典的羰基化反應是不飽和化合物(烯烴、炔烴)、鹵代烴和甲醇等在親核試劑的存在下與CO反應生成應用廣泛的有機含氧化合物,如圖2所示。
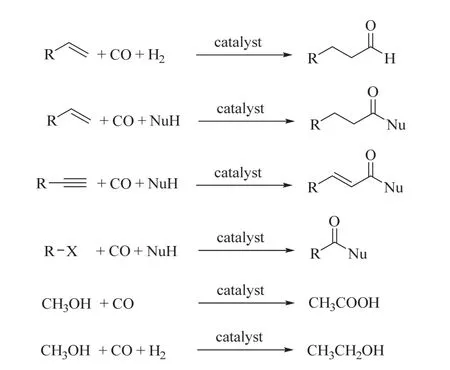
圖2 CO參與的羰基化反應Fig. 2 The conventional carbonylation with CO.
因此,近年來,人們尤為感興趣的是使用CO2/H2替代CO合成高附加值化學品23,以期在未來工業生產中實現CO2作為主要含碳化學品的來源。Leitner等在Science的Perspective中就描述了CO2/H2替代CO的前景,將CO2/H2形容為“初見不鐘情”,這是因為CO2是“不情愿的”搭檔(熱力學穩定和動力學惰性),但在特殊催化劑(主要為過渡金屬絡合物)的“撮合”下,實現了CO2/H2“再見鐘情”,參與到高價值化合物的合成中(如圖3所示)24。
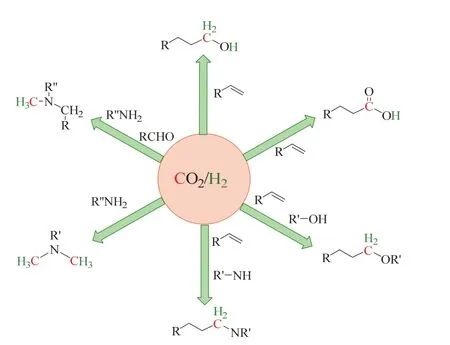
圖3 Leitner等描述的CO2/H2替代CO的前景Fig. 3 The prospect of CO2/H2 as CO surrogate described by Leitner et al.
過渡金屬催化CO2/H2的組合為替代CO提供了一種可能,為合成高價值的化學品創造了基礎,潛力巨大。本綜述對近年來CO2/H2參與的羰基化研究進展進行了簡要的總結和評述。在此,我們將CO2/H2看成類似魂斗羅一樣不斷闖關的搭檔,每闖過一個關卡即成功實現了一種羰基化反應,在這些關卡中會出現不同的障礙:氫化/氫解、反應條件苛刻、一般都涉及多個競爭反應、化學選擇性低、區域選擇性差、貴金屬用量過高或催化性能低等,而化學家們通過給兩位“魂斗羅”裝備不同的“道具”(如鹵代鹽、離子液體、膦配體等)成功克服了通向目標產物道路上的“障礙和敵人”。
2 CO2/H2參與的烯烴羰基化反應
烯烴的官能團化是當今化學工業的重要基礎。除了聚合和氧化外,使用CO進行烯烴羰基化反應是工業生產醛、醇、羧酸、酯、胺等高附加值和精細化學品的主要技術(如圖4所示),對烯烴的高效羰基化也是有機化學研究中的重點課題和挑戰之一25,26。
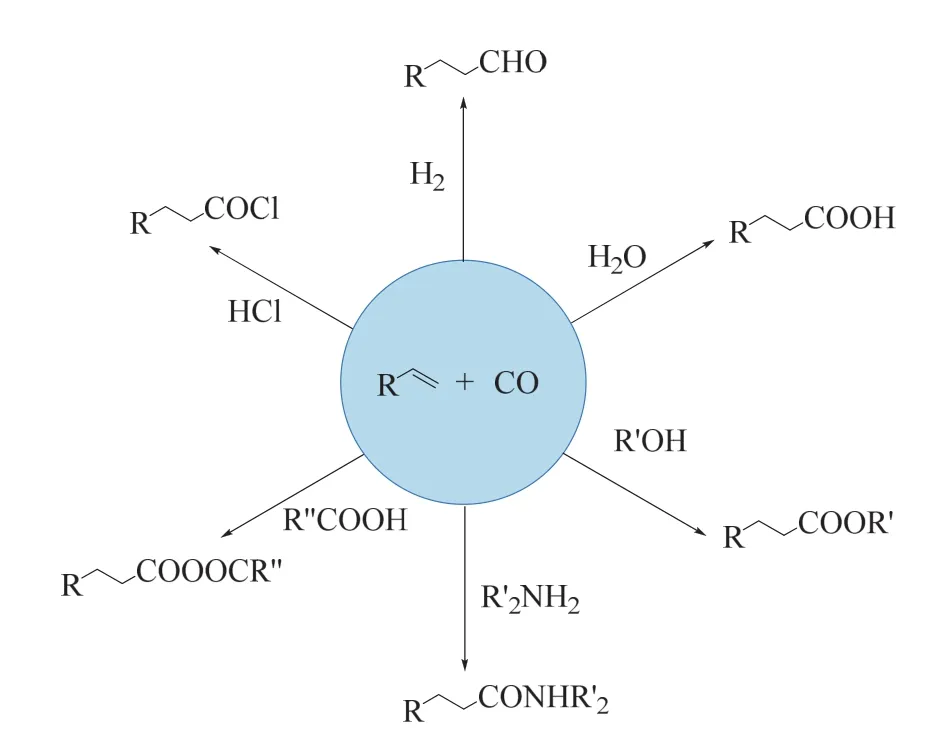
圖4 CO參與的烯烴羰基化反應Fig. 4 The carbonylation of alkene with CO.
眾多研究者對CO2/H2作CO替代品參與烯烴羰基化反應進行了廣泛地研究和探索27,在過渡金屬催化下成功實現了CO2/H2參與烯烴羰基化反應生成醛、醇、羧酸、酯和胺等28。在此,我們綜述了以CO2/H2為CO替代品的烯烴羰基化反應的研究進展。根據已有的研究,在CO2/H2參與的烯烴羰基化反應體系中目前主要有四種策略(如圖5):①釕催化的RWGS/烯烴氫甲酰化/氫化的多步串聯反應生成醇,若將還原劑H2替代為醇,則可生成產物酯;②負載在二氧化鈦(TiO2)上的金(Au)為代表的多相催化體系成功實現了多相RWGS/乙烯氫甲酰化/氫化制丙醇的反應;③銠催化的RWGS/烯烴氫羧化串聯反應生成高級羧酸;④ CO2被聚甲基氫硅氧烷(PMHS)還原為CO,隨后經銠催化的烯烴氫甲酰化選擇性生成醛。這些催化體系通過巧妙的設計實現了CO生成和烯烴羰基化的高效耦合,同時不同的催化體系也存在著各自的優劣勢。我們通過對不同烯烴羰化反應類型的分析,闡述該領域的研究進展。
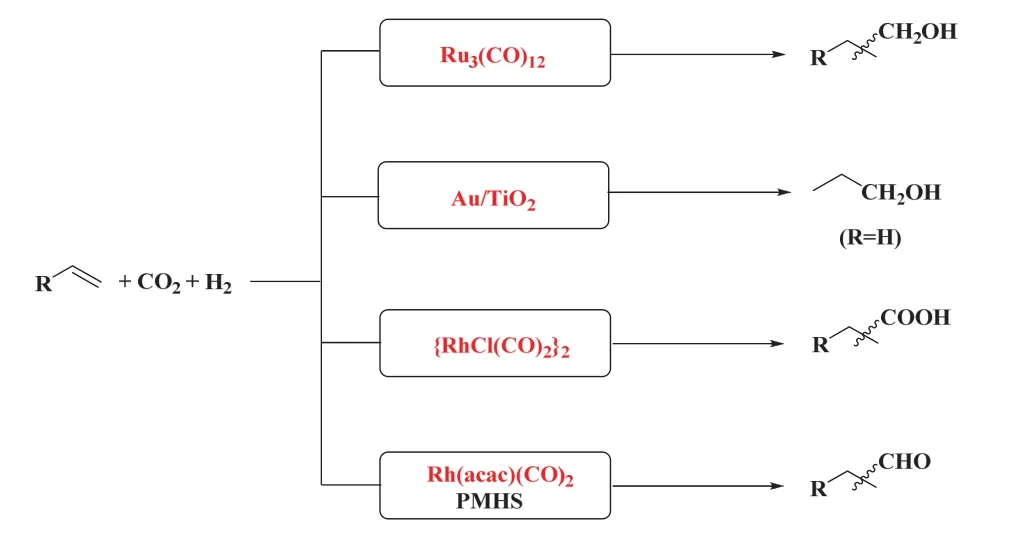
圖5 CO2/H2參與的烯烴羰基化反應的策略Fig. 5 Strategies for carbonylation of alkenes with CO2/H2.
2.1 CO2/H2參與的烯烴氫甲酰化/氫化制醇
1993年,Sasaki課題組首次報道了均相催化RWGS,在CO2/H2氛圍下,以十二羰基三釕(Ru3(CO)12)為催化劑,KI為添加劑,反應機理如圖6a所示29。首先,CO2插入Ru―H鍵中(A),在H2輔助下脫去一分子水,生成羰基Ru絡合物(B),隨后B脫除CO,脫羰后的Ru進一步加氫生成Ru―H鍵,催化劑循環再生。但該體系中羰基Ru絡合物(B)同時會生成副產物CH3OH和CH4等。次年,同課題組在上述催化體系的基礎上,引入LiCl或雙(三苯基正膦基)氯化銨([PPN]Cl)為添加劑,不僅使CO2高效選擇性轉化為CO,同時成功抑制了CH4和CH3OH的生成,反應溫度也顯著下降(圖6b所示)30,為利用原位生成的CO參與羰基化反應提供了可能。基于Ru3(CO)12催化RWGS的特性,同一課題組于2000年將該體系拓展至烯烴氫甲酰化反應中,首次實現了CO2/H2參與的RWGS/烯烴氫甲酰化/氫化制醇的反應(如圖7a所示)31。該催化體系使用四核羰基釕(H4Ru4(CO)12)為催化劑,LiCl為添加劑,反應在140 °C、CO2/H2(4/4 MPa)條件下進行。CO2首先經歷RWGS生成CO;在H2和原位生成的CO參與下,烯烴發生氫甲酰化反應生成醛。但在高溫條件以及具有顯著氫化傾向的Ru存在下,反應產物不能停留在醛,而是進一步氫化生成醇,最終環己基甲醇收率可達88% (以環己烯為模板底物)。這項開創性的工作引起了廣泛關注,眾多科研工作者在此基礎上深入研究了CO2/H2參與烯烴制醇的反應,以期開發出CO2為羰源的適宜工業化的烯烴氫甲酰化的新型催化劑。
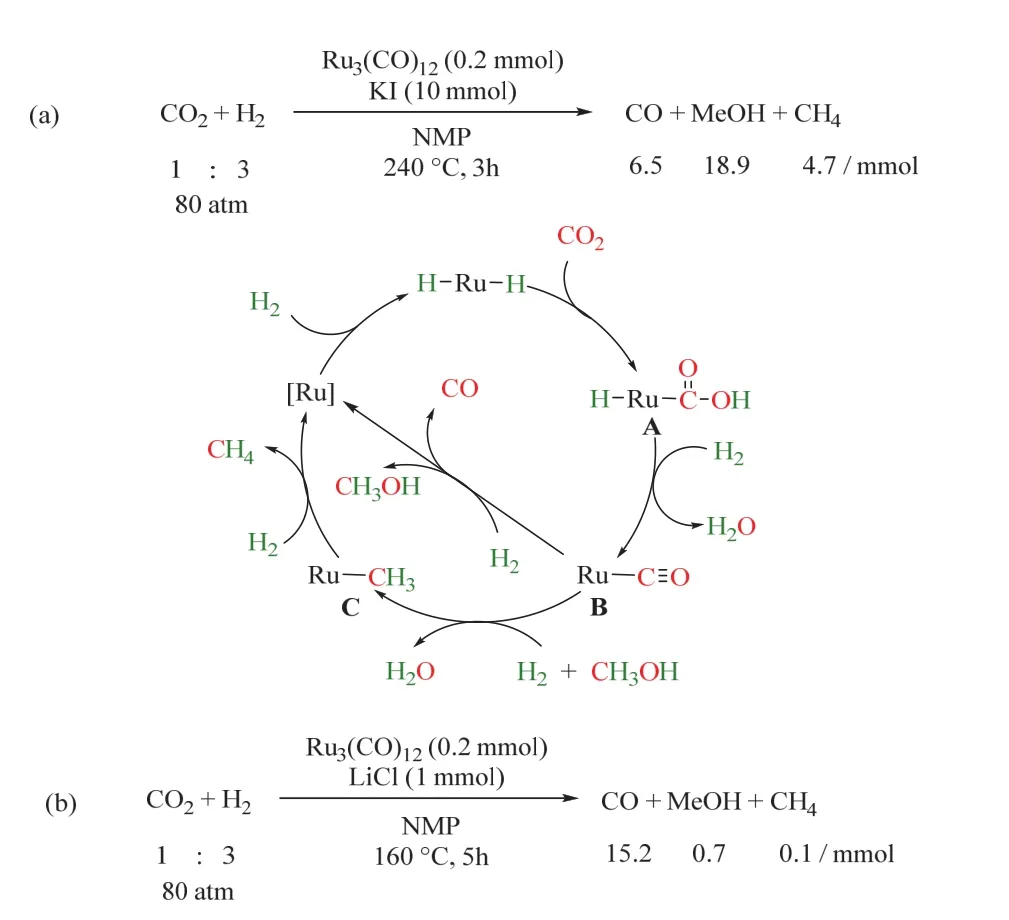
圖6 Ru3(CO)12催化CO2逆水煤氣變換為CO機理圖Fig. 6 A plausible reaction mechanism of RWGSR.
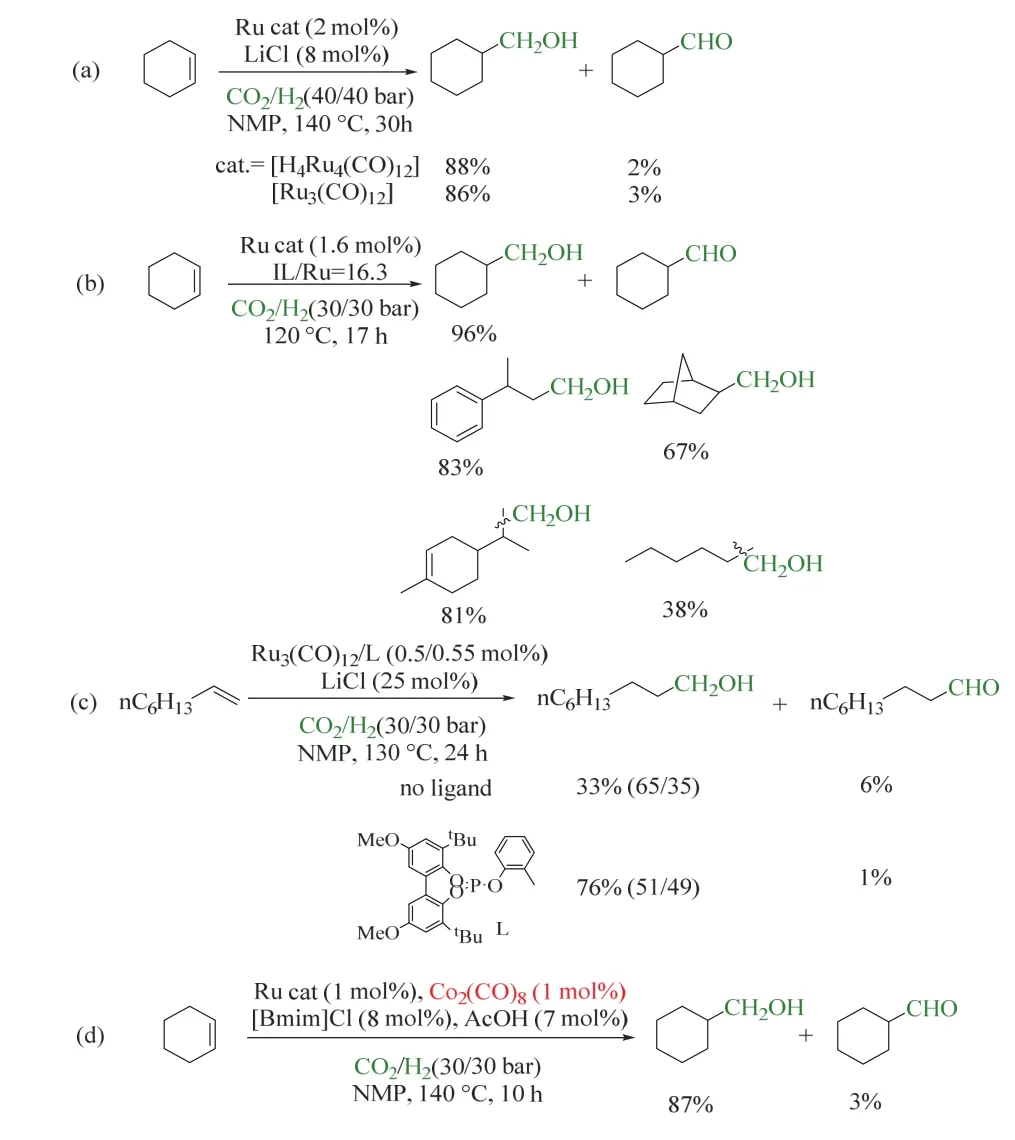
圖7 Ru催化的CO2/H2參與的烯烴羰基化反應制醇Fig. 7 Ru-catalyzed alkene carbonylation to produce alcohols with CO2/H2.
后續研究發現鹵素添加劑對氫甲酰化反應也有著顯著影響32-34,當沒有添加劑鹵代鹽時,氫甲酰化反應不發生,僅得到烯烴氫化產物。金屬催化劑前體中羰基不可或缺,不含羰基的常見釕金屬鹽無法催化這類反應。同時N或P配體的引入降低了反應性能,可能的原因是配體抑制了CO的生成。在相同的反應條件下,與傳統的CO羰基化相比,CO2表現出相似甚至更好的性能。其反應循環機理如圖8所示34,外圈經歷RWGS生成CO,ESIMS表明該過程是由四核陰離子物種催化的。首先Cl-脫除[H2Ru4(CO)12]2-中的質子生成不含氫的絡合物[Ru4(CO)12]4-,這是該過程的關鍵步驟;隨后CO2配位生成[Ru4(CO)12(CO2)]4-,質子親核進攻進一步的把配位的CO2轉化成CO配體,形成[Ru4(CO)13]4-;后者在H2輔助下脫去CO,同時再生為[H2Ru4(CO)12]2-。原位生成的CO隨后參與內圈的氫甲酰化反應。烯烴首先與催化活性中心[RuCl3(CO)3]-生成烷基釕絡合物[R’RuCl2(CO)3],隨后CO插入,在HCl輔助下脫去醛,催化劑[RuCl3(CO)3]-循環再生;最后,醛被Ru催化氫化生成醇。由圖可以看出,鹵素離子的主要作用是脫除質子,從而生成具有反應活性的催化中心羰基釕陰離子[Ru4(CO)12]4-。
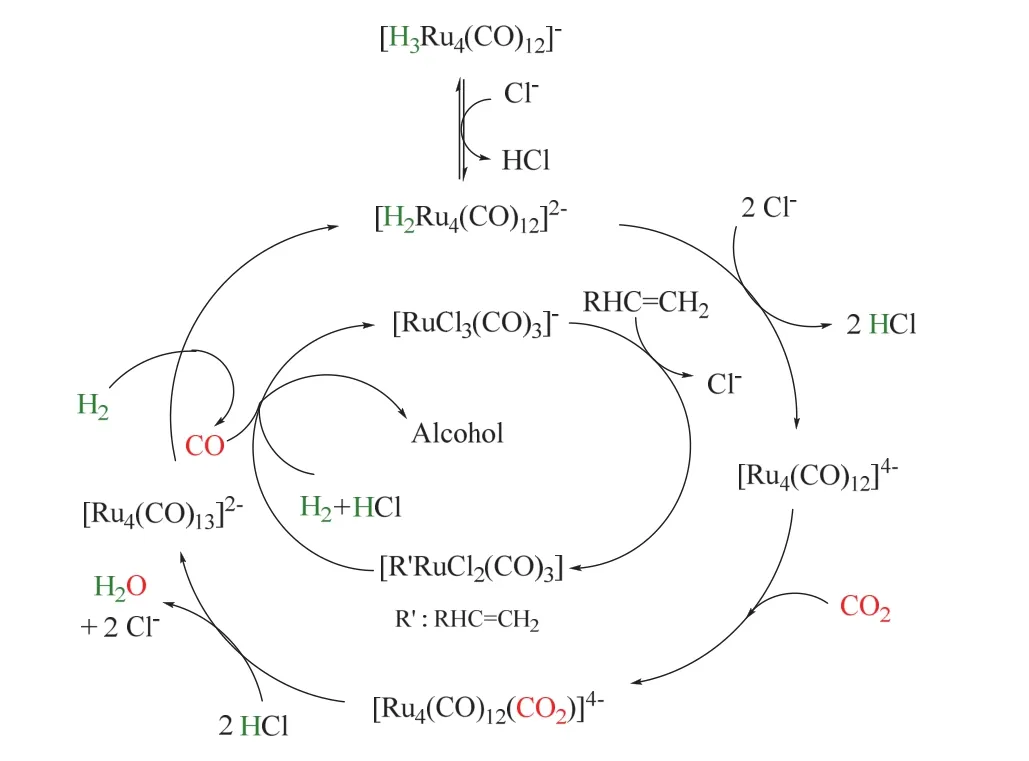
圖8 Ru催化CO2/H2參與烯烴羰基化制醇的反應機理Fig. 8 The mechanism of Ru-catalyzed hydroformylation/reduction of alkenes with CO2/H2.
離子液體是指室溫或低溫下為液體的熔融鹽,由有機陽離子和無機/有機陰離子(如鹵素離子)組成;離子液體具有極低的蒸氣壓,這有效消除了揮發性溶劑對環境和人體的危害。基于其各方面優異的特性,離子液體在CO2/H2參與的氫甲酰化反應中可同時充當添加劑和綠色溶劑。Tominaga以1-己烯為底物,在[Bmim]Cl/甲苯或[Bmim][Cl +NTf2](Bmim:1-丁基-3-甲基咪唑;NTf2:二(三氟甲基磺酰)酰亞胺)的混合溶劑中獲得了超過80%的庚醇35。Dupont課題組則進一步將離子液體完全替代鹵素鹽和溶劑的使用,同時H3PO4等酸的引入有利于Ru催化的RWGS,在相對溫和的反應條件下,環己烯和多種2,2-二取代烯烴均獲得較高的醇收率(如圖7b所示)36。
基于上述的研究,Beller課題組考察了多種亞膦酸酯配體,通過具有較大空間位阻的亞膦酸酯配體與Ru3(CO)12相配合,實現了CO2/H2參與的脂肪端烯和內烯烴的氫甲酰化反應,在相對溫和的條件下獲得醇產物(如圖7c所示)。膦配體的引入有效抑制了烯烴的加氫,提高了對應醇的收率37。
羰基釕體系催化RWGS/烯烴氫甲酰化/氫化的串聯反應雖然得到了有價值的醇,但催化劑中貴金屬釕的用量較高,成本較大,這限制了該類反應的進一步發展。在前述工作的基礎上,何林課題組構建了釕鈷雙金屬催化體系(圖7d),其中釕基催化體系實現了RWGS/醛氫化,而Co2(CO)8則催化烯烴氫甲酰化,這有效降低了貴金屬釕的用量。理論計算表明,體系中酸的引入便于Ru-COOH中間體的脫羥,從而推動了RWGS,使該體系在生成醇的反應中表現出良好的性能。適用烯烴包括多種來自生物質和石油工業的廢料,均可通過該策略轉化為有價值的醇,這也同時實現了工業廢料的增值和CO2利用38。
考慮到釕的稀缺性、高成本以及均相體系中催化劑回收難等問題,發展多相催化體系吸引了很多研究者的興趣。一方面多相催化劑具備易回收和循環使用的優勢,另一方面多相催化劑也在RWGS和傳統烯烴氫甲酰化中表現優異,因此,多相催化的CO2/H2參與烯烴氫甲酰化在理論上是可行的。
雖然CO2/H2參與多相氫甲酰化研究的策略與均相類似,均為RWGS和氫甲酰化反應的串聯,但依然存在一些挑戰。首先是反應溫度不匹配:受熱力學限制,多相中RWGS所需溫度較高(一般高于300 °C),而傳統氫甲酰化的溫度較低(100 °C左右),實現兩個反應的耦合具有一定難度;其次在反應過程中副反應較多:如烯烴加氫、CO2甲烷化等。因此,多相催化體系的創制具有較大挑戰性。
Kondratenko課題組在CO2/H2為合成氣替代品的多相乙烯氫甲酰化制丙醇的轉化中做出了一系列開創性工作。該課題組首次實現Au/TiO2體系催化的CO2/H2參與乙烯氫甲酰化制丙醇的反應39,在2 MPa (CO2/H2/C2H4= 1 : 1 : 1)進料氣和200 °C條件下,摻雜K的Au/TiO2催化RWGS生成的CO全部參與到烯烴氫甲酰化反應中,丙醇收率為2.4%,而不摻雜K的Au/TiO2體系丙醇收率僅為0.7%。機理研究發現Au吸附的CO2通過RWGS還原為CO,原位生成的CO 100%選擇性的參與乙烯氫甲酰化反應生成丙醛,丙醛進一步加氫生成丙醇。研究表明通過Au沉積方法和K添加劑的引入可以對Au的顆粒尺寸進行調節,且反應的活性和選擇性與Au的顆粒尺寸密切相關。K的摻雜不僅能調控Au的尺寸,而且促進吸附氫物種的形成,推動了CH3CH2CO加氫生成丙醇40。然而,K添加劑的摻雜也降低了CO2的轉化率,CO2被強吸附在堿性位點上生成離子型碳酸鹽(如碳酸鉀),而離子型碳酸鹽分解為氫甲酰化所需要的CO則非常緩慢。考慮到總體上堿性添加劑K對該反應的積極作用,該課題組考察了堿金屬Cs對催化性能的影響41。研究表明Cs與K類似,可明顯影響產物的選擇性,最終引入Cs的催化體系可獲得2.9%的丙醇。實驗表征顯示Cs存在于載體和Au顆粒之間,這有利于Auδ+的穩定,促進了乙烯的羰基化反應速率,進而提高了丙醇的選擇性。
研究發現載體類型對反應性能也有很大影響,該課題組制備了K-Au/TiO2-r (金紅石)和Au/TiO2-a (銳鈦礦)等催化體系并用于探究不同類型的TiO2載體對反應性能的影響。對比發現KAu/TiO2-r體系更加利于丙醇的生成(詳見表1)40,丙醇收率可達3.1%。金屬和載體之間的相互作用對金粒徑尺寸和分布的影響可能是影響反應的主要原因。該課題組進一步對比了K-Au/TiO2和KAu/SiO2的催化性能(如表1條目2和條目5所示)42,研究發現在TiO2負載的催化劑得到的醇收率比SiO2負載的催化劑更高。實驗表征發現TiO2載體表面的碳酸鹽和甲酸鹽物種不會抑制CO2還原和氫甲酰化反應的發生。反應過程中Auδ+轉變為活性位點Au0,乙烯在活性Au0位點被活化,與CO和H2快速反應,最終生成丙醇。而K-Au/SiO2體系更有利于CO2的轉化,且隨著K含量的提高,SiO2載體促進Au對乙烯和氫氣的活化,并顯著抑制烯烴加氫。這可能是因為K在SiO2中部分溶解形成硅酸鉀層,從而抑制了對RWGS不利的碳酸鉀的形成(如前所述,碳酸鉀和CO2的結合力更強,使CO2難以被還原為CO) (如圖9所示)43。
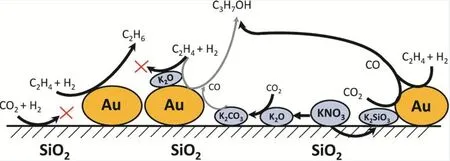
圖9 載體和添加劑對金納米粒子在CO2/H2和C2H4合成丙醇中的活性和選擇性的影響Fig. 9 Effect of support and promoter on activity and selectivity of gold nanoparticles in propanol synthesis from CO2/H2 and C2H4.
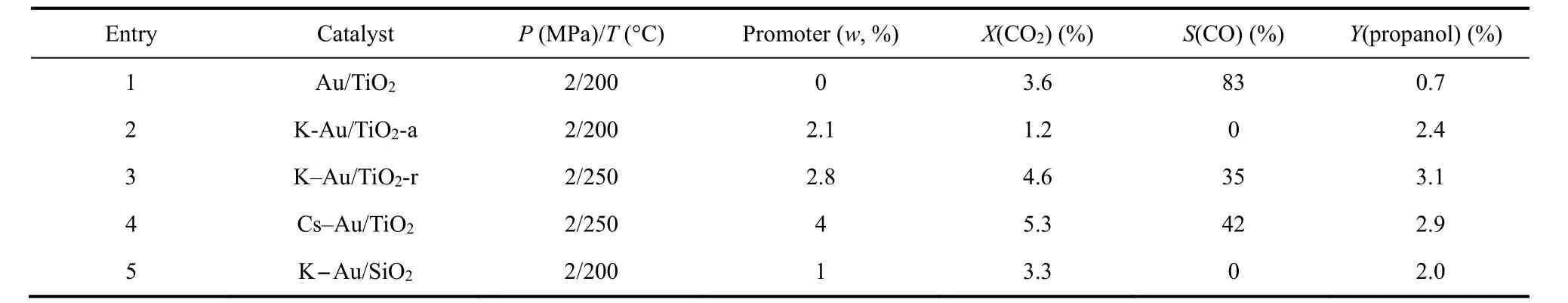
表1 Au基多相催化CO2/H2參與的乙烯氫甲酰化性能比較Table 1 Performance comparison for hydroformylation of ethylene with CO2/H2 catalyzed by Au-based heterogeneous catalysts.
Kondratenko課題組開創了不同于均相羰基釕的Au/TiO2多相催化體系,首次實現了在CO2/H2條件下乙烯到丙醇的轉化,闡明了載體和添加劑對催化性能的影響,但在該類催化體系中丙醇的收率較低,反應條件苛刻,后續的探究需進一步提高醇收率和使反應條件溫和化。
綜上,CO2/H2參與的烯烴氫甲酰化/氫化制醇的反應已經取得了長足的發展,CO2/H2在各種“道具”的幫助下成功突破了眾多“關卡”,如以羰基釕為催化劑,添加劑由最初的鹵代鹽發展到更綠色環保的離子液體。在溫和反應條件下,實現了多種環烯烴、2,2-二取代烯烴等的高效轉化成醇,膦配體與羰基釕的成功結合也進一步實現了脂肪端烯和內烯烴的高效轉化,同時釕鈷雙金屬體系的開發降低了貴金屬的使用。值得關注的是,Au/TiO2多相催化體系的開發成功實現了乙烯到丙醇的轉化,拓寬了該類反應的催化劑類型,為催化劑的循環再利用提供了基礎。雖然通過離子液體和配體分別促進了脂肪端烯的醇收率和區域選擇性的一定提升,但獲得高化學/區域選擇性的醇/醛這一有挑戰性的“關卡”仍需進一步探索。
2.2 CO2/H2參與的烯烴及其衍生物的氫羧基化制高級羧酸
在CO2作為羧基源的烯烴還原羧化生成高級羧酸的研究中,如圖10a所示,還原劑主要使用超化學計量的乙基鋅、鎂試劑、金屬錳等有機金屬試劑或有機硅試劑44-47,這些試劑一般來說價格昂貴,且對空氣/水敏感,這限制了該催化方法的應用。H2是一種清潔、廉價易得的還原劑,基于CO2/H2的烯烴還原羧化制高級羧酸是更具前景和挑戰性的(圖10b)。
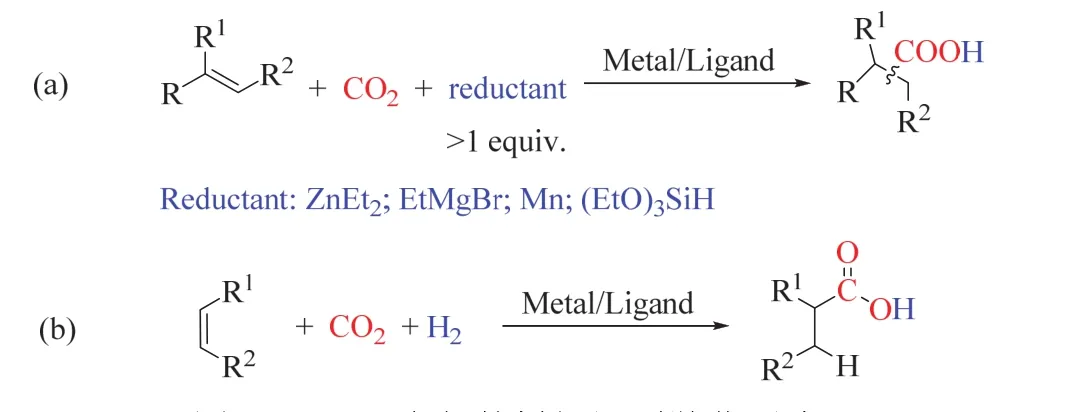
圖10 CO2參與的烯烴還原羧化反應Fig. 10 Reduction carboxylation of alkene with CO2。
Leitner課題組提出了從CO2、H2和烯烴直接合成高級羧酸的催化策略(圖11)48。該策略以[RhCl(CO)2]2為金屬前體,PPh3為配體,碘甲烷(CH3I)和對甲苯磺酸(PTSA)的引入促進了氫羧化反應的進行,多種烯烴在180 °C、CO2/H2(60/10 bar,1 bar = 0.1 MPa)的條件下反應生成增一個碳的羧酸,其收率最高可達92%。實驗研究顯示單齒膦配體通常適合用于該反應體系,而多齒膦配體在該反應中則完全抑制了羧酸的形成,可能的原因是多齒膦配體抑制了RWGS的進行。對反應歷程的探究表明,乙酸環己酯可能為反應活性最高的中間物,醇也因此可充當有效的底物,其轉化率和產物分布與對應的烯烴基本一致。同位素實驗表明,羧酸基團并非由CO2分子直接嵌入構筑,而是經歷了CO和H2O作為活性物種的傳統氫羧化途徑。整個轉變可能通過Rh催化的RWGS反應、原位生成的CO和H2O隨后參與烯烴的氫羧化反應。該反應把逆水煤氣變換中的產物H2O也利用起來,實現了100%的原子利用率。
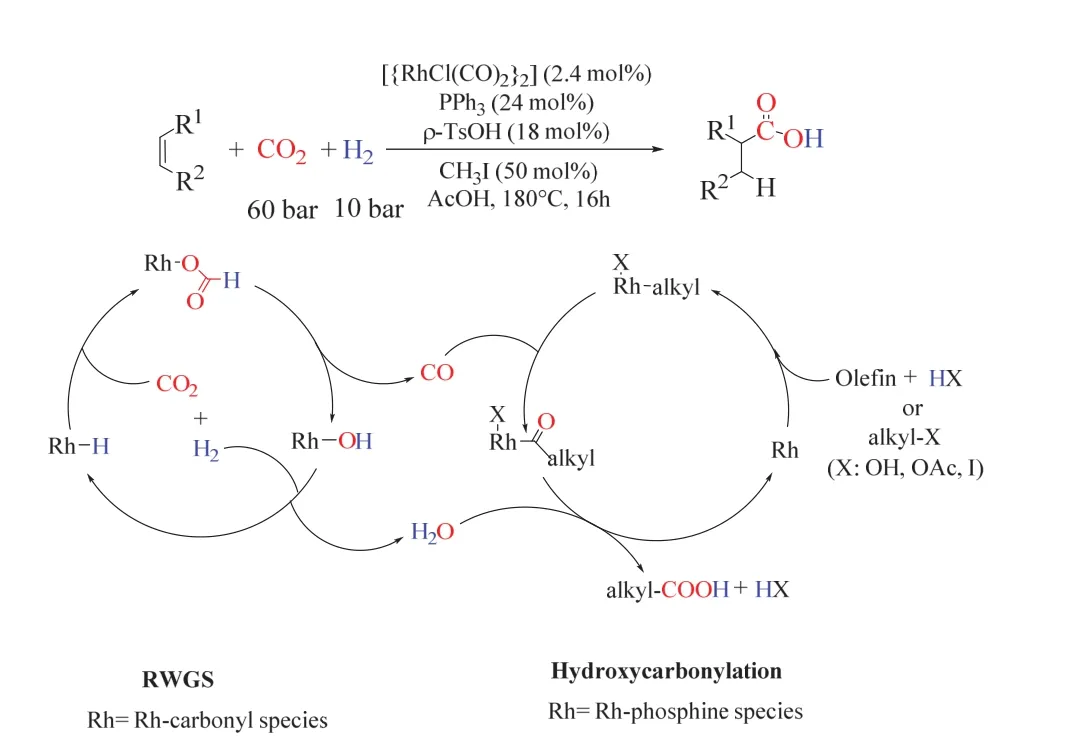
圖11 CO2為C1源的烯烴氫羧化反應生成高級羧酸Fig. 11 CO2 as a C1 building block for the formation of carboxylic acids by hydrocarboxylation.
該策略提供了一種新的以CO2為C1源合成高級羧酸的方法,實現了從CO2/H2組合出發制備高級羧酸的“關卡”的突破。近期韓布興課題組報道的Ir催化CO2/H2參與的醚制高級羧酸。在該催化體系下醚會首先轉化為烯烴,隨后與原位生成的CO發生羰基化反應。因此,我們將醚視做烯烴的衍生物。
一般來說,醚的反應活性低于烯烴或醇,且沒有作為底物和CO2/H2制備高級羧酸的前例。韓布興課題組首次實現了由醚、CO2和H2合成高級羧酸,在170 °C、5/2 MPa CO2/ H2氛圍下,以LiI為促進劑的IrI4體系高效催化該轉化(如圖12所示),獲得了優異收率的高級羧酸49。該催化體系具有良好的底物普適性,各種醚如環醚、直鏈烷基醚、支鏈烷基醚和芳香醚等都能被高效轉化為相應的高級羧酸。機理研究表明,底物醚被快速選擇性地轉化為烯烴,這是該策略順利進行的關鍵步驟,烯烴與I-進一步轉化為烷基碘化物,RWGS原位產生的CO插入到烷基碘化物中,隨后被原位形成的水還原消除,最終生成增一個碳的羧酸。該方法中底物醚相對烯烴更為廉價易得,具有較大的應用前景。
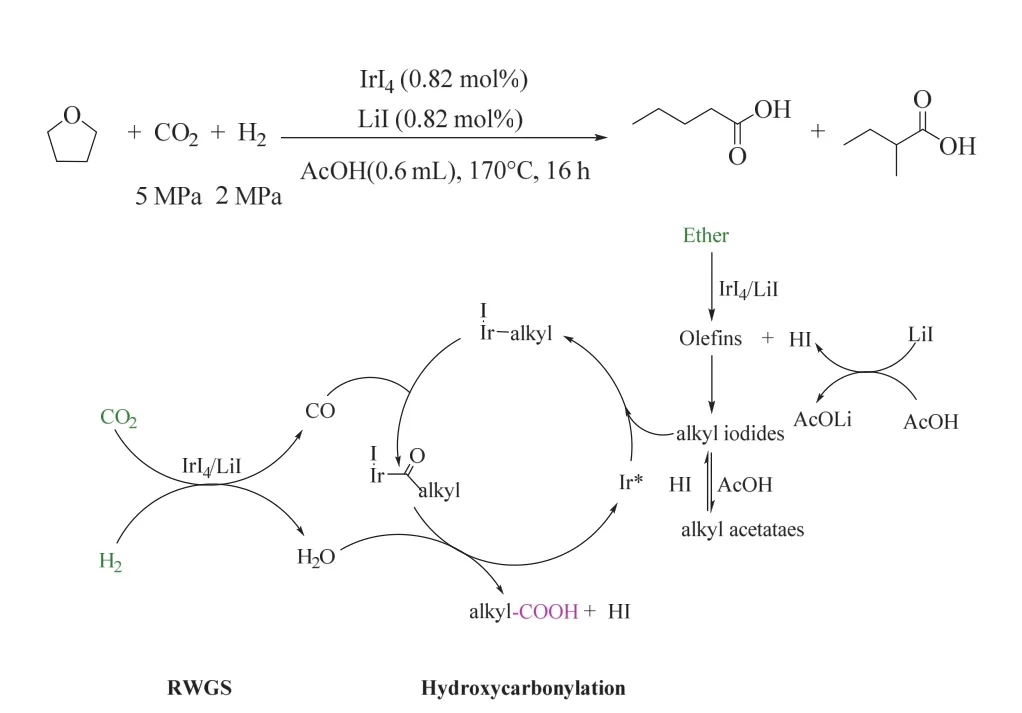
圖12 IrI4催化CO2/H2參與的醚羰基化生成羧酸的反應49Fig. 12 Ir-catalyzed synthesis of carboxylic acid from ether, CO2/H2 49.
綜上,在CO2/H2參與烯烴及其衍生物醚的氫羧化生成高級羧酸的反應過程中,Rh和Ir均成功實現了RWGS和烯烴氫羧化反應的高效耦合,得到了增一個碳的高級羧酸。該類反應實現了原子的高效利用,但催化性能還有待提升,如需要相對苛刻的反應條件以及較大劑量的添加劑。因此,反應條件溫和化,并減少添加劑的使用等是下一步研究重點。
2.3 CO2/ROH參與的烯烴氫酯化制酯
烯烴羰基化反應不僅可提供醛、醇,還可提供工業中重要的聚合物中間體羧酸酯(這類轉化也稱為氫酯化反應),如目前工業上生產聚甲基丙烯酸甲酯(有機玻璃)的工藝便是基于鈀催化的乙烯氫酯化反應50-52。
Beller課題組發展了一種新穎的烯烴氫酯化反應53, 以CO2作C1源,MeOH為還原劑,避免使用敏感或昂貴的還原劑。該催化體系以Ru3(CO)12為催化劑,離子液體[Bmim]Cl為添加劑,在相對溫和的條件下實現了烯烴氫酯化反應,最終得到產物酯。脂肪烯烴和芳香烯烴在該催化體系下均可高效轉化為對應的羧酸酯,如從乙烯出發生成工業中重要的丙酸甲酯;此外,乙醇和苯甲醇替代甲醇在該體系中也能實現很好的轉化。作者通過同位素標記和控制實驗對反應機理進行了探究,發現CO2是主要的羰基來源,同時CH3OH也可充當羰基源,基于此,作者提出了幾種可能的途徑,分別經歷CO和甲酸甲酯中間體(圖13)。該策略首次實現了從CO2/醇出發合成羧酸酯,這種轉化進一步拓寬了CO2作為C1源參與形成C―C鍵的催化領域。
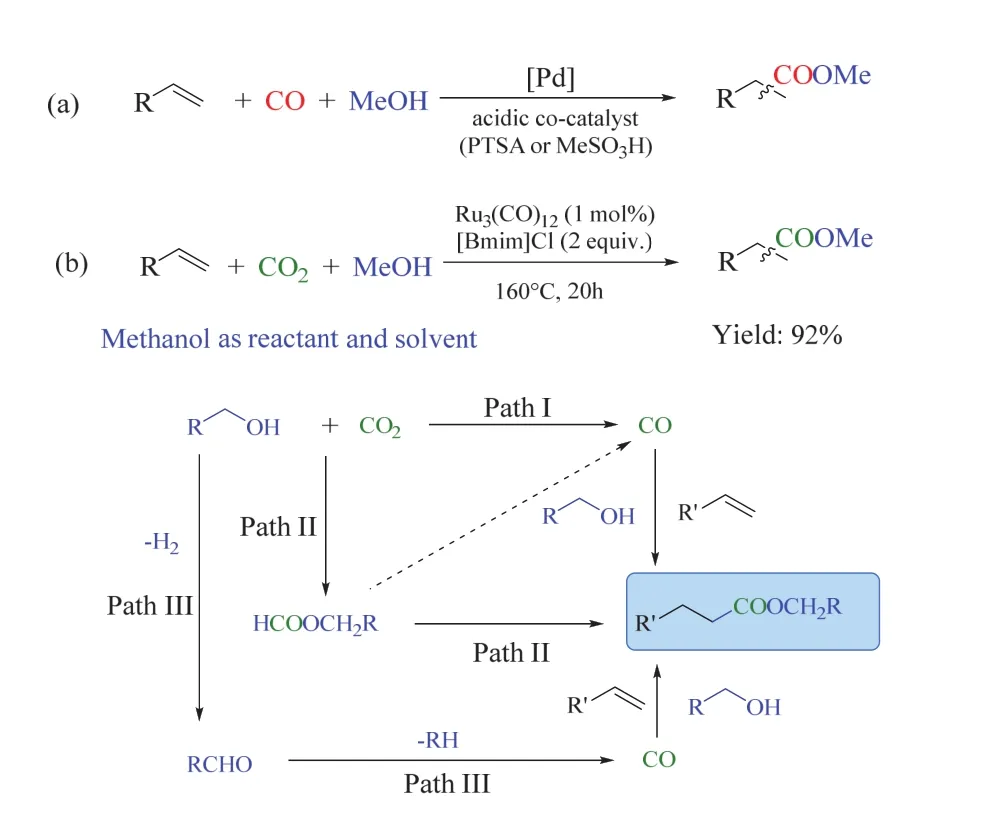
圖13 釕催化CO2/醇參與烯烴氫酯化反應53Fig. 13 Ru-catalysed alkoxycarbonylation of alkenes with CO2/alcohols 53.
上述氫酯化過程包含CO2還原和烯烴氫酯化兩個步驟,使用單一催化劑同時高效催化該串聯反應是困難的,這是因為各反應對催化劑的要求是不同的。基于這一考慮,何林課題組發展了釕-鈷雙金屬協同催化CO2還原/烯烴氫酯化的體系(圖14)54。該雙金屬體系表現出良好的催化性能和可重復使用性,與之前單釕的催化體系相比,二元催化體系可有效地減少貴金屬和離子液體添加劑的使用。機理研究表明釕催化劑主要參與CO2還原,環己烯的氫酯化則依賴于鈷催化劑。密度泛函理論(DFT)計算從能量的角度闡明了[Ru(CO)3Cl2]2緩慢分解形成活性釕催化劑以及烯烴氫化的發生。
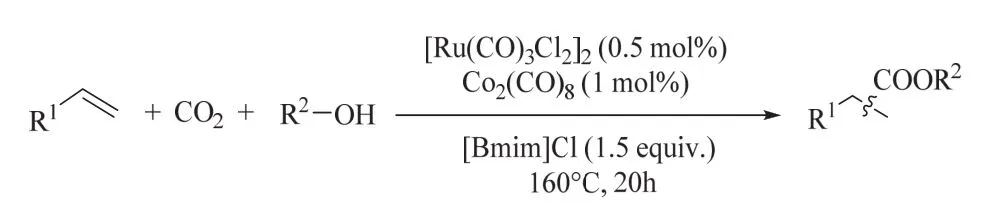
圖14 釕-鈷雙金屬協同催化CO2/醇參與烯烴氫酯化Fig. 14 Alkoxycarbonylation of alkenes with CO2/alcohols by a reusable heterobimetallic ruthenium cobalt catalytic system.
羰基釕催化的CO2/ROH參與的烯烴氫酯化反應開辟了新的合成路線,從烯烴、CO2/醇得到了之前未能實現的酯的產物,但該體系釕用量較高,反應條件較苛刻。因此,開發更加溫和、高效的催化體系仍是令人期待的。
2.4 CO2/H2參與的烯烴氫甲酰化/還原胺化制胺
與其他烯烴官能團化相比,胺的合成在有機化學中也引起越來越多的關注55。除了作為大宗化學品,由于其優異的生物活性,仲胺和叔胺也可作藥物發現方面重要的靶標。本節基于前述Ru催化CO2/H2參與烯烴氫甲酰化,研究者向該體系中引入伯胺或仲胺,原位生成的醛與胺還原胺化,分別生成仲胺或叔胺。值得一提的是,本文主要涉及經典的C―C鍵構筑的羰基化反應,而如CO2/H2參與的胺羰基化/胺甲基化等直接形成C―N鍵的羰基化反應也得到廣泛報道23,56,但不在本節討論范圍內。
Eilbracht等報道了首例Ru催化的RWGS/烯烴氫甲酰化/還原胺化多步串聯反應,該催化體系以Ru3(CO)12為催化劑,LiCl為添加劑,芐基三乙基氯化銨(BTAC)為相轉移劑,在CO2/H2(20/60 bar),160 °C條件下反應5天后,能以35%-98%的收率得到多種期望的仲或叔胺(圖15)57。該體系順利進行的關鍵在于LiCl作為添加劑抑制烷烴、醇、甲酰胺等副產物的形成,相轉移催化劑的加入提高了LiCl的溶解度,并對整體選擇性產生了積極影響。在該策略下,醛加氫到醇的過程被抑制,醛被胺捕獲發生不可逆地縮合并還原,最終得到胺的產物。
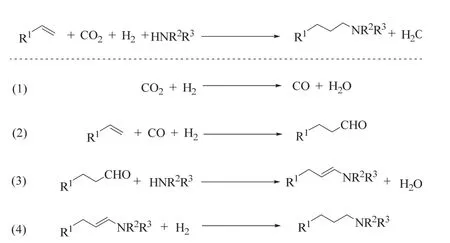
圖15 釕催化CO2/H2和胺參與的烯烴氫胺甲基化Fig. 15 Ru-catalyzed hydroaminomethylation of alkenes with CO2/H2 and amines.
在上述工作的基礎上,Dupont課題組將Ru3(CO)12溶解在離子液體中,同樣高效的實現了該串聯轉化:(a) RWGS,(b) 烯烴氫甲酰化,(c)醛的還原胺化。在120 °C,CO2/H2(60 bar,1 : 1)條件下,烯烴與伯胺或仲胺(烯烴/胺= 1 : 1)在Ru3(CO)12催化下反應,反應時間縮短至36 h,烯烴轉化率高達99%,胺的選擇性高達96%58。該體系比之前報道的體系催化效率更高,同時降低了貴金屬的使用量,并將反應條件進一步溫和化。
盡管反應路徑復雜,CO2/H2參與的烯烴氫甲酰化/還原胺化仍實現了仲胺或叔胺的高效制備,后續可在更具普適性的烯烴和胺底物、更趨溫和的反應條件以及更短的反應時間等上實現該“關卡”的突破。
2.5 CO2/H2參與的烯烴氫甲酰化制醛
如前所述,CO2/H2參與烯烴羰基化可獲得醇、羧酸、酯、胺等產物,其反應策略主要基于過渡金屬催化RWGS得到CO的反應路徑,但RWGS所需的高溫和釕的過度氫化能力使烯烴氫甲酰化中目標產物不能停留在醛,而是進一步氫化生成醇。而中間體醛作為大宗化學品在工業中具有非常重要的地位。為了選擇性得到醛的產物,丁奎嶺課題組經過巧妙的策略設計,以Rh(acac)(CO)2和螺環骨架的亞膦酸酯配體為催化劑,并加入額外的還原劑PMHS,最終實現了溫和條件下(100 °C,5/20 bar CO2/H2)的烯烴氫甲酰化反應,獲得高化學/區域選擇性的直鏈醛,其中醛收率可達70%,直異比可達10 : 1 (圖16)59。作者通過控制實驗證實了CO的生成來源于CO2與PMHS反應,而非RWGS,這避免了RWGS所需的苛刻條件,同時溫和的條件有利于提高醛的直異比。CO2被PMHS還原優先于醛的還原以及Rh/亞磷酸酯催化體系的弱氫化能力是反應得以停留在醛的關鍵。
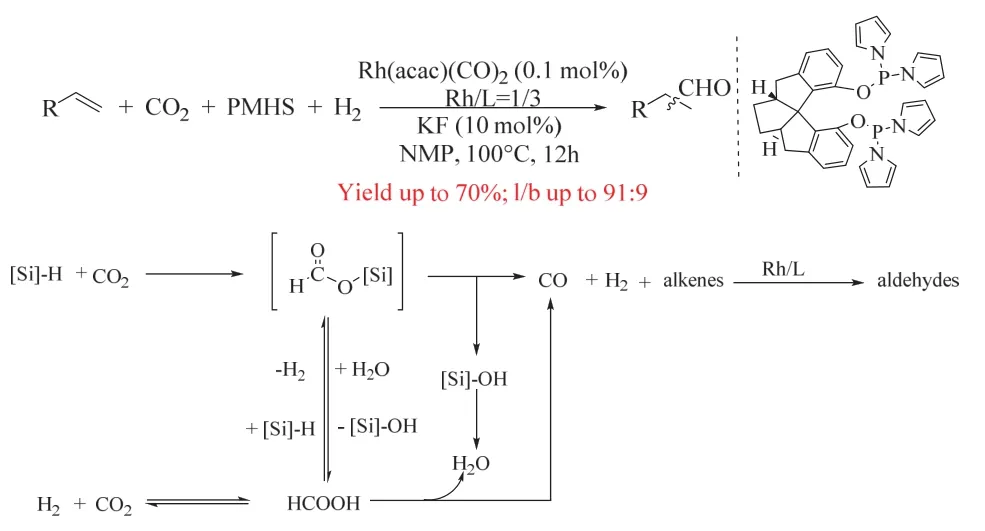
圖16 銠催化CO2和氫硅烷參與的烯烴氫甲酰化反應Fig. 16 Rhodium complex-catalyzed hydroformylation of alkenes with CO2 and hydrosilane.
通過引入額外的還原劑PMHS,CO2/H2參與的烯烴氫甲酰化可選擇性獲得直鏈醛,這為CO2/H2參與的烯烴羰基化反應提供了一種新的思路。然而,額外還原劑PMHS的使用造成了部分底物的損失和產物分離的困難。因此,以H2為唯一還原劑的CO2/H2作合成氣替代品的烯烴氫甲酰化反應仍是令人期待的。
最近,陳經廣課題組通過串聯的固定床成功將CO2、乙烷脫氫重整和氫甲酰化高效耦合,實現了丙醛和丙醇的生成60。該策略采用多相Fe3Ni1/CeO2體系催化CO2輔助乙烷的脫氫重整,獲得乙烯、CO和H2;而RhCox/MCM-41催化劑則順利促進乙烯的氫甲酰化反應選擇性生成C3含氧化物(丙醛和丙醇) (如圖17所示)。該策略在無外加還原劑的存在下實現了CO2/C2H6兩種惰性、廉價分子轉化為高價值化學品醛和醇,為CO2參與的羰基化反應提供了新的思路。
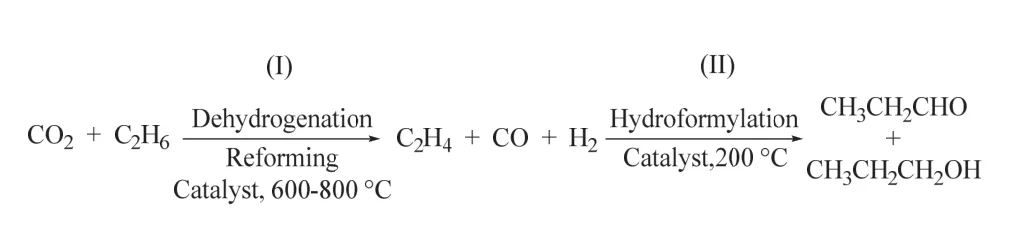
圖17 多相催化CO2和乙烷反應制丙醛/丙醇Fig. 17 The conversion of CO2 and ethane to propionaldehyde/propanol catalyzed by heterogeneous catalyst.
3 CO2/H2參與的鹵代烴羰基化反應
芳香醛是C―C、C―N和C―S偶聯反應中的重要中間體,在材料、藥品、農藥和農用化學品的合成中有著廣泛的應用61。合成芳香醛的傳統方法包括羧酸或酯還原,Gattermann-Koch、Reimer-Tiemann、Duff和Vilsmeier反應等62,這些方法通常存在某些固有缺陷,如收率低、選擇性差、需要有毒氣體和苛刻條件等。基于此,利用CO或CO替代品作為C1源,從芳香鹵化物直接甲酰化合成芳香醛的轉化逐漸發展起來。如圖18a-c所示,CO或甲醛,甲酸等CO替代品均能在鈀催化下高效生成芳香醛63-65。這一進展也促進了使用CO2代替CO進行芳香鹵化物甲酰化的探索。劉志敏課題組在CO2參與的芳香鹵代物羰基化反應中做出了開創性的工作,首次以CO2為C1源,在氫硅烷PMHS和堿1,8-二氮雜二環十一碳-7-烯(DBU)的存在下,實現了Pd催化芳香碘/溴化物66,67直接甲酰化生成芳香醛(圖18d)。
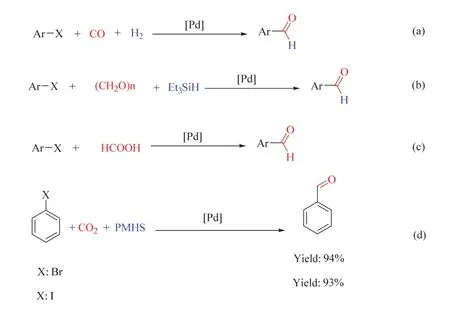
圖18 鈀催化CO及其替代品的芳基鹵代烴甲酰化反應Fig. 18 Palladium-catalyzed formylation of aryl halides with CO and its derivatives.
在上述工作的基礎上,同一課題組首次使用CO2/H2參與芳香鹵化物的甲酰化轉化(圖19)68。該催化體系由RhI3(適用于芳香碘化物)或RhI3/Pd(dppp)Cl2(適用于芳香溴化物)和PPh3組成,乙酸酐(Ac2O)為脫氧試劑,并以三乙胺(Et3N)為堿性添加劑,該體系在100 °C下以優異的收率得到芳香醛,并表現出良好的官能團耐受性和廣泛的底物適用性。控制實驗表明反應經歷了三個串聯步驟,分別是在Et3N輔助下CO2加氫生成HCOOH(或HCOOH-NEt3加合物),其在Ac2O存在下分解、釋放CO,原位生成的CO與芳香鹵化物經歷傳統的甲酰化反應生成芳香醛。
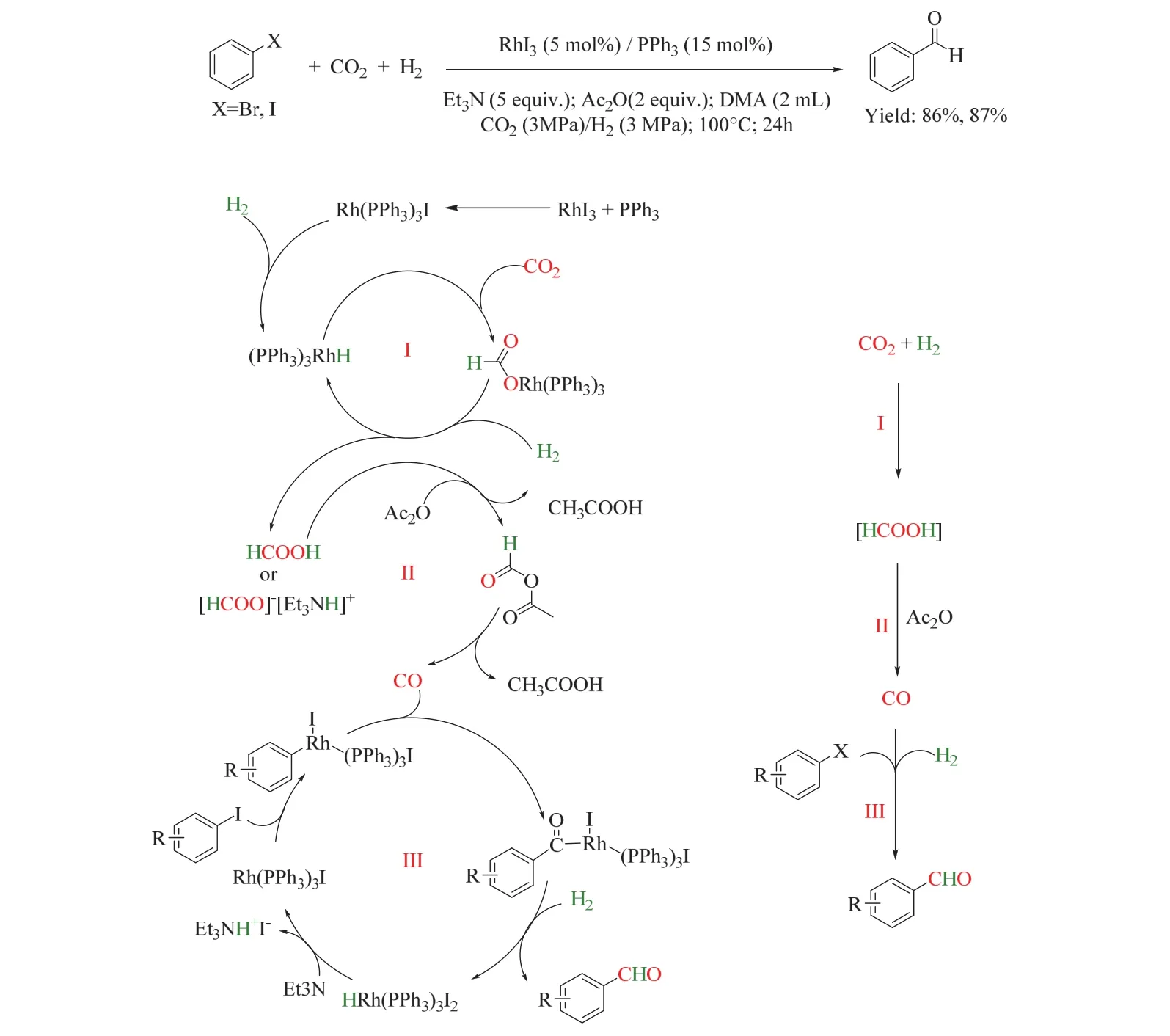
圖19 銠催化的CO2/H2參與的芳香鹵代烴甲酰化Fig. 19 Rhodium-catalyzed formylation of aryl halides with CO2 and H2.
這種不同于RWGS路徑的、經歷甲酸中間體的多步串聯的反應路徑實現了溫和條件下CO2氫化間接轉化為CO,為CO2/H2實現更多的羰基化反應提供了兼容的可能性。
4 CO2/H2參與的甲醇及其衍生物的羰基化反應
乙酸是一類重要的基礎大宗化學品,可用作合成乙酸乙烯酯單體和乙酸酐的原料,目前工業中主要通過甲醇羰基化的方法生產,以來自于化石燃料的CO為羰基源69-71。利用甲醇及其衍生物(如甲氧基醚等富含OCH3的化合物)和CO2/H2合成乙酸等化學品對綠色可持續發展具有重要意義,但也具有很大挑戰性72,73。
乙醇作為一種重要的大宗化學品,被廣泛用作溶劑、食品、醫藥、農藥、有機原料等,同時作為可替代燃料在當前的基礎能源設施中發揮著日益重要的作用。目前,乙醇主要是通過石油生成的乙烯催化水合或玉米、甘蔗等食物發酵生產。因此,以CO2和H2為原料合成乙醇具有重要意義,但這仍然是一個巨大的挑戰,因為它涉及到CO2加氫的同時形成C―C鍵73,74。使用CO2合成乙醇的報道大多局限于高溫下CO2加氫,該路線中,CO2通常與H2反應生成高活性C1中間體,包括CO、CH3*(吸附態的CH3)或CH3OH,這些中間體經過C―C鍵偶聯形成C2+產物,例如乙醇和高級醇75,76。由于CO2氫化原位生成C1中間體和碳鏈增長同時發生,因此產物分布通常較寬,乙醇在醇類產物中的占比較低。為了提高乙醇的選擇性,引入特定底物與CO2和H2反應是一種可行的方法。例如以甲醇、CO2和H2為起始原料,乙醇是唯一醇類產物,其他甲醇衍生物如甲醛、二甲醚(DME)等,也可作為底物與CO2/H2反應選擇性生成乙醇。
本節總結了近年來CO2/H2參與的甲醇或其衍生物羰基化制乙酸、乙醇的報道,其中韓布興課題組在該領域做出了開創性的工作。
4.1 CO2/H2參與的甲醇及其衍生物羰基化制乙酸的反應
利用可再生的、廉價的CO2合成乙酸具有重要意義,但同時也具有很大挑戰性。目前CO2參與乙酸合成的路線存在明顯的局限,如選擇性低、催化劑活性低、反應溫度高、使用昂貴或有毒的反應物等。例如,以CO2和CH4為原料合成乙酸受限于熱力學限制,往往需要很高的溫度,且乙酸的產率較低77。而使用CH3I、CO2和H2為反應物時,可獲得選擇性10.7%的乙酸,但CH3I高毒且價格昂貴。因此,實現CO2高效、高選擇性生成乙酸是令人期待的78。
韓布興課題組報道了甲醇、CO2和H2合成乙酸的新型反應路徑(圖20)79。該催化體系以Ru3(CO)12/Rh2(OAc)4為催化劑,咪唑為配體,LiI為促進劑,在180 °C以上可高收率的獲得乙酸,其轉化頻率(TOF)值達到30.8 h-1,基于甲醇的乙酸收率高達77%,且催化劑經過5次循環后,催化性能幾乎保持不變。體系中配體咪唑對催化劑的高催化活性、穩定性和選擇性起著關鍵作用。令人意外的是,乙酸生成的反應不通過CO參與的反應路徑,CO在反應的氣體樣品中幾乎檢測不到。隨后,作者通過同位素實驗證實CO2是通過直接插入參與的羰基化反應。反應機理如圖所示:甲醇與LiI首先形成CH3I (步驟1),隨后CH3I與活性Rh*生成CH3Rh*I (步驟2),CO2直接插入CH3Rh*I中形成CH3COORh*I (步驟3),該過程中Rh催化乙酸的生成。在H2的存在下活性Ru物種(Ru*)促進CH3COORh*I還原消除生成乙酸(步驟4),最后,原位生成的LiOH和HI中和形成LiI和H2O (步驟5)。所有的催化物種再生后進行下一個循環。
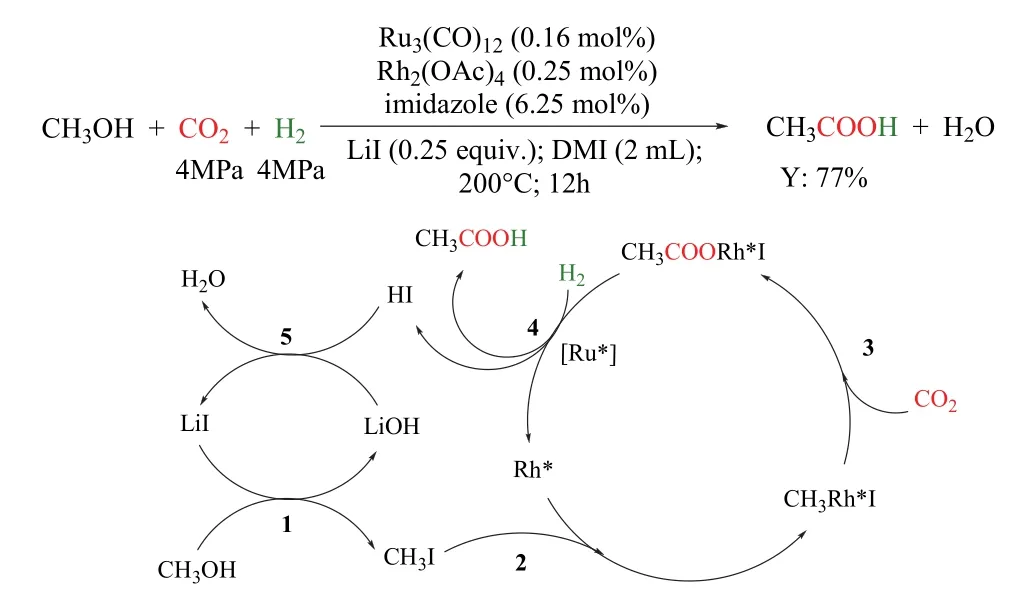
圖20 Ru-Rh雙金屬催化的CO2/H2參與的甲醇羰基化制乙酸79Fig. 20 Synthesis of acetic acid via methanol hydrocarboxylation with CO2 and H2 by Ru-Rh bimetallic catalyst 79.
在此工作的基礎上,同一課題組將雙金屬體系發展為單金屬Rh2(CO)4Cl2/4-甲基咪唑(4-MI)/LiI體系(如圖21所示)。該反應在150 °C時乙酸開始形成,在180 °C時乙酸收率最高可達81.8%,此時TOF高達26.2 h-1。與雙金屬體系相比,該催化體系更簡單,且在溫和的條件下效率更高80。
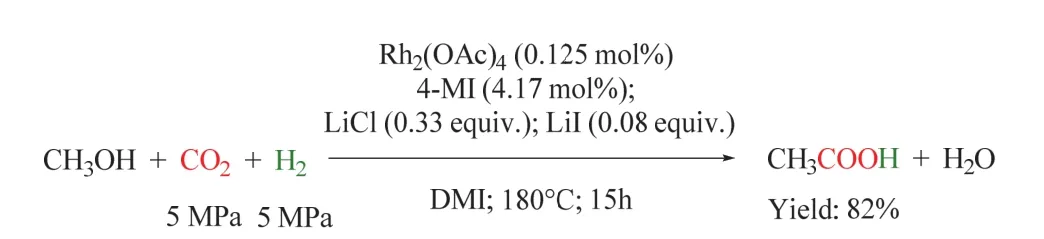
圖21 Rh催化的CO2/H2參與的甲醇制羧酸反應Fig. 21 Synthesis of carboxylic acid from methanol with CO2/H2 by Rh catalyst.
眾所周知,木質素是一類擁有豐富含氧基團(例如OCH3)的生物高聚物,其中的OCH3可充當甲醇衍生物,參與到羰基化反應中。基于此,劉志敏課題組使用前述類似的雙金屬催化體系,實現了苯甲醚(木質素中最簡單的平臺化合物)在CO2/H2條件下合成乙酸,其中苯甲醚可提供甲基生成CH3I。在180 °C,3/3 MPa CO2/H2條件下時,乙酸的收率可達94% (圖22),值得注意的是,離子液體(如[Bmim]Cl)充當溶劑并促進了催化活性中心的形成,機理探究表明[BMIm]+穩定的RhI3和I-絡合的Ru(CO)n(n = 1-4)是形成乙酸的關鍵活性催化物81。其后,韓布興課題組以離子液體1-己基-3-甲基咪唑四氟硼酸鹽(HMimBF4)為添加劑,在相對溫和的條件下也實現了苯甲醚/木質素與CO2、H2生成乙酸的轉化82。
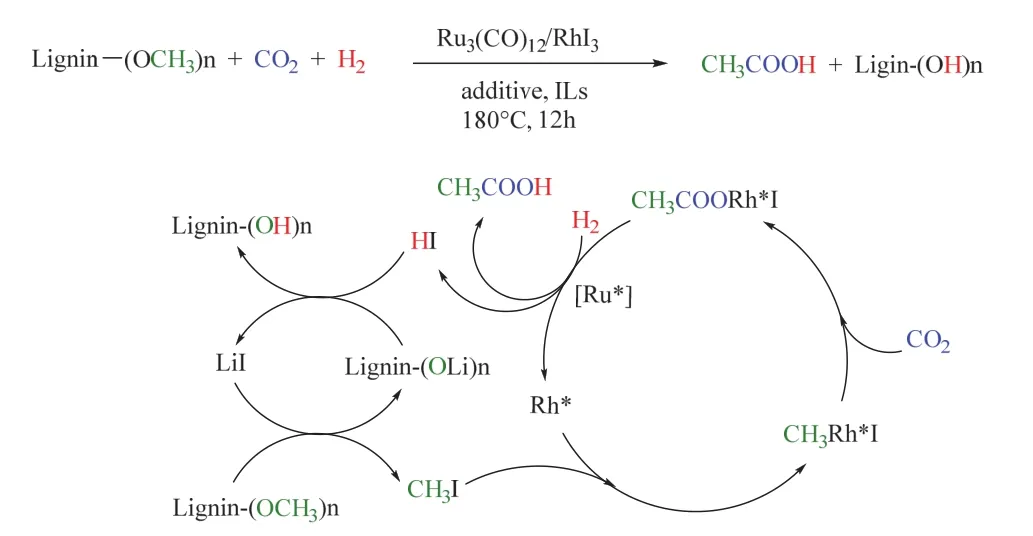
圖22 Ru-Rh雙金屬催化CO2/H2參與木質素羰基化制乙酸Fig. 22 Ru-Rh bimetal-catalyzed carbonylation of lignin to acetic acid with CO2/H2.
綜上,CO2/H2參與甲醇及其衍生物羰基化制乙酸的反應近年來得到快速發展,Rh/Ru雙金屬體系首次實現CO2直接插入的羰基化反應生成乙酸,揭示了CO2/H2參與羰基化反應的新的反應路徑。在保證乙酸選擇性和收率的基礎上,通過配體篩選(咪唑到4-甲基咪唑),添加劑(鹵素鹽到離子液體)和反應條件的優化,實現了單金屬Rh高效催化該反應,提高了催化性能,降低了貴金屬的使用,同時實現了苯甲醚/木質素等富含OCH3的甲醇衍生物與CO2、H2生成乙酸的轉化,拓寬了該類反應的底物范圍。但該體系銠用量較高,反應條件較苛刻。后續可在更溫和的反應條件以及更多含OCH2-R類型的化合物(如甲醛、乙醇等)上實現多種羧酸“關卡”的突破。
4.2 CO2/H2參與甲醇及其衍生物羰基化制乙醇的反應
在多數情況下,C2+產品,如乙醇、乙酸、烯烴和液體燃料等相對C1產品用途更為廣泛。但CO2和H2高效、高選擇性地直接合成C2+產物相對困難,因為這同時涉及到可控的CO2加氫和C―C鍵形成,這在基礎科學中仍然是一個挑戰73,83-86。目前,CO2加氫直接制乙醇的反應溫度普遍較高,反應活性和選擇性較低。基于此,引入特定底物如甲醇或其衍生物(如甲醛、二甲醚等)與CO2/H2反應合成乙醇是提高催化活性和選擇性的可行方法。從反應機理的角度考慮,反應一般被認為經歷了源自RWGS的CO插入生成乙醛的過程,因此可被歸到羰基化反應之列。
1998年Sasaki課題組首次報道了CO2/H2參與的甲醇同系化制乙醇的反應(如圖23所示),該體系以Ru-Co雙金屬為催化劑,兩種金屬間存在協同作用,乙醇的收率受所加添加劑鹵代鹽的顯著影響,其中LiI性能最佳。10 mmol甲醇在180 °C和2/10 MPa CO2/H2條件下反應15 h,乙醇收率最高可達3.2 mmol,乙醇選擇性為34.2 C-mol% (代表以碳摩爾數計的產率)87。作者推測甲醇被來自于CO2的CO所同系化生成乙醇。盡管該體系的催化性能有待提高,但它為高選擇性制乙醇開辟了新的途徑。
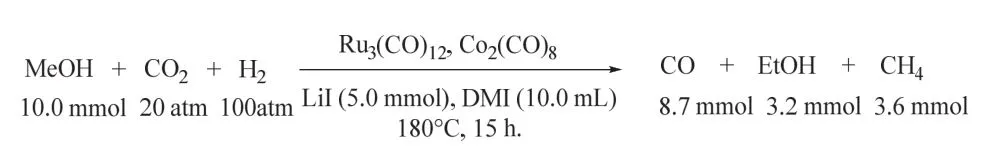
圖23 釕-鈷雙金屬催化CO2/H2參與甲醇體系化的反應Fig. 23 Methanol homologation using CO2/H2 catalyzed by ruthenium-cobalt bimetallic complex system.
基于上述工作,韓布興課題組在2017年報道了多聚甲醛(可視作甲醇的衍生物)為反應底物、CO2和H2參與合成乙醇的反應88。該體系以LiI作為促進劑,在大于140 °C,CO2/H2(3/5 MPa)條件下,Ru-Co雙金屬催化劑可高效協同催化該轉化,基于金屬Ru的乙醇TOF值最高可達17.9 h-1,乙醇在總產物中的選擇性達到50.9 C-mol% (如圖24所示)。此外,該催化體系能多次重復使用,在5個循環后乙醇的總轉化數(TON)超過805。機理研究表明該轉化由三個串聯反應組成,分別是:甲醛氫化生成甲醇,RWGS得到CO,甲醇被CO同系化,最終生成乙醇。基于對機理的深入探究,同一課題組把引入的特定底物從甲醛拓展到二甲醚89、甲醇90,91和芳基甲基醚/木質素92,在相似的催化體系下同樣順利實現了CO2/H2參與的乙醇合成。其中以甲醇為特定底物時,實現了單一金屬Ru促進的乙醇生成90,且使用離子液體作為溶劑,擴展了這類反應的溶劑范圍。
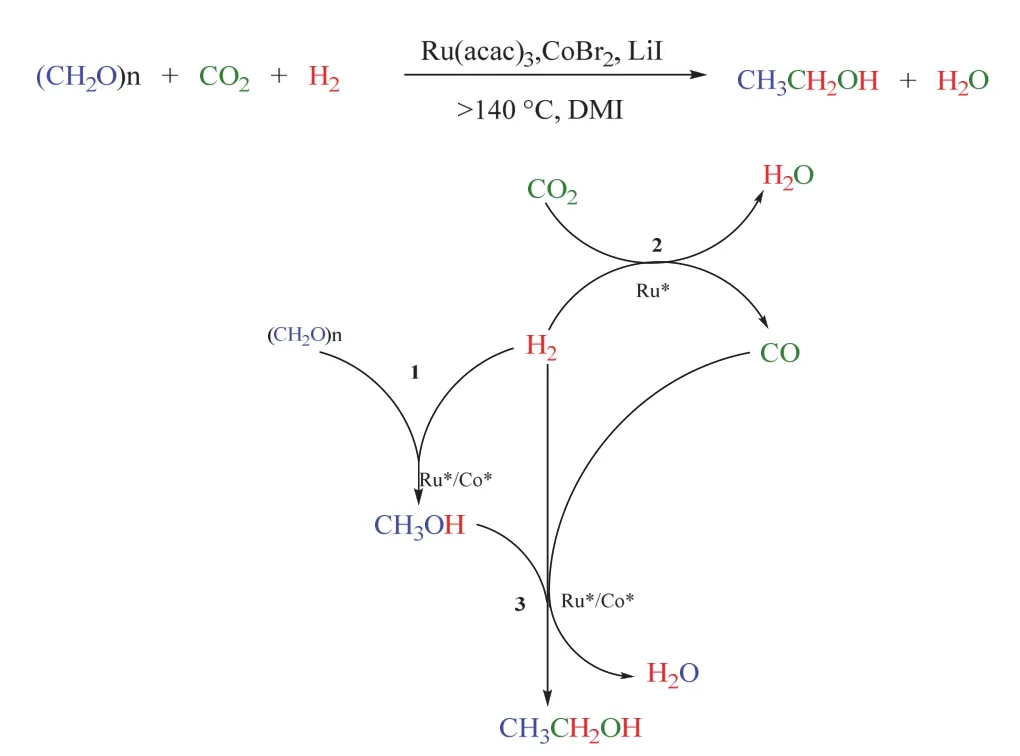
圖24 Ru-Co催化的多聚甲醛、CO2和H2合成乙醇Fig. 24 Ru-Co catalyzed synthesis of ethanol from paraformaldehyde, CO2 and H2.
綜上,CO2/H2參與甲醇及其衍生物羰基化制乙醇的反應得到進一步發展,利用可多次重復使用的Ru-Co雙金屬高效協同催化該轉化,實現了RWGS/甲醇同系化制乙醇,通過添加劑篩選和反應條件的優化進一步提高了乙醇的選擇性,也實現了單一金屬Ru催化的乙醇生成。同時該類轉化的底物也由甲醇拓展到甲醛、二甲醚和芳基甲基醚等,拓寬了該類轉化的底物范圍。后續可在更溫和的反應條件以及更多含OCH2-R類型的化合物上實現多元醇“關卡”的突破。
5 結論與展望
CO2作為一種豐富、廉價的C1源被廣泛用于生產多種高價值的化學品,H2作為最有前景的還原劑之一,與CO2的組合在過渡金屬的催化下實現了多種羰基化反應,可獲得諸如醛、醇、羧酸、酯、胺等高附加值化學品。在這類反應中,過渡金屬(釕、銠、銥)催化的RWGS生成CO是此類轉化的關鍵步驟,隨后原位生成的CO隨后參與到羰基化反應中。通過向催化體系中引入鹵代鹽或離子液體、膦配體、非貴金屬鈷等手段一定程度上抑制了烯烴氫化、提高了區域選擇性和反應效率,降低了貴金屬用量。同時多相Au基催化劑的開發為催化劑設計提供了一條新的思路。此外,甲醇及其衍生物的羰基化高化學選擇性、高收率的生成乙醇進一步拓展了CO2/H2參與的羰基化反應的領域。除了需高溫高壓的RWGS反應路徑,CO2加氫生成甲酸、隨后分解為CO的反應路徑也被初步探索,實現了溫和條件下CO2間接轉化為CO,并與芳香鹵代物的甲酰化高效耦合,實現了CO2/H2參與構筑了甲酰基。與CO2氫化為CO參與羰基化的反應路徑相比,CO2直接參與羰基化的反應路徑豐富了CO2/H2參與羰基化的模式,實現了CO2作為羰基源的甲醇制乙酸的反應。這些CO2/H2參與的不同類型的羰基化反應和模式拓展了CO2資源化利用的途徑,獲得高附加值的大宗或精細化學品,促進了綠色化學的發展。
盡管CO2/H2參與的羰基化反應已經有較多且深入的研究,但這些催化體系距離高化學選擇性、高區域選擇性以及潛在的工業化還有很長一段路要走,主要原因如下:
(1) RWGS路徑和CO2直接插入的反應路徑常需高溫高壓,反應條件苛刻,這限制了配體發揮作用,導致了較低的催化性能和化學/區域選擇性,如烯烴氫甲酰化/氫化所得到的醇直異比整體都較差。而CO2加氫到甲酸、甲酸分解為CO的過程中需要消耗(超)化學計量的乙酸酐,這些均限制了CO2/H2參與羰基化反應的進一步發展。
(2) 絕大多數CO2/H2參與的羰基化反應常需要含量較高的添加劑以抑制副反應的發生。
(3) 貴金屬用量過高,催化劑的高效回收再利用需要整體解決方案。
基于上述存在的問題,開發新穎的催化體系和羰基化反應類型仍是重要的研究方向。為了實現這些“關卡”,完成這些挑戰,以下幾個方面值得進一步深入探究:
(1) 實現溫和條件下的RWGS,并與后續的過渡金屬/配體催化的傳統羰基化相耦合,實現高化學/區域選擇性的羰基化。
(2) 實現CO2加氫到甲酸、甲酸無需當量添加劑的消耗即分解為CO、CO參與羰基化反應的高效串聯耦合。有研究表明,甲酸可在含有內置堿的配體作用下選擇性分解成CO,這為該多步串聯反應的設想提供了可行性。
(3) 減少該類反應貴金屬的使用量,提高金屬催化劑的催化性能,也可通過非貴金屬部分或全部替代貴金屬來催化CO2/H2參與的羰基化反應。
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11

- 物理化學學報的其它文章
- Understanding the Role of Cu/ZnO lnteraction in CO2 Hydrogenation to Methanol
- Electrocatalytic CO2 Reduction to Ethylene over CeO2-Supported Cu Nanoparticles: Effect of Exposed Facets of CeO2
- Controlling the Global Mean Temperature by Decarbonization
- Cu@UiO-66 Derived Cu+-ZrO2 Interfacial Sites for Efficient CO2 Hydrogenation to Methanol
- 二氧化碳電還原反應的理論研究
- 離子液體介導CO2化學轉化研究進展