金屬助催化劑對(duì)介孔Ni基催化劑加氫提質(zhì)木質(zhì)素的影響
2021-06-07 05:45:28丁世磊李福威趙婷婷李志霞熊德元
應(yīng)用化工 2021年5期
關(guān)鍵詞:催化劑
丁世磊,李福威,趙婷婷,李志霞,熊德元
(1.廣西中醫(yī)藥大學(xué) 藥學(xué)院,廣西 南寧 530200;2.廣西大學(xué) 化學(xué)化工學(xué)院,廣西 南寧 530004)
木質(zhì)素是植物細(xì)胞壁的重要組成部分,是一種重要的可再生資源,通過(guò)催化加氫精制可以制備燃料、高附加值的醇類產(chǎn)物或者酚類原料,是高效使用木質(zhì)素資源的一條新途徑[1-2],但木質(zhì)素化學(xué)性質(zhì)穩(wěn)定,催化加氫精制過(guò)程中所需反應(yīng)條件較高[3-5],開(kāi)發(fā)新型高效催化劑是提高木質(zhì)素精制效率的重要途徑。愈創(chuàng)木酚作為木質(zhì)素液化的重要產(chǎn)物,其分子中含有酚羥基和甲氧基兩個(gè)含氧官能團(tuán)及一個(gè)苯環(huán),具有較強(qiáng)的自聚能力[6-7]。本文擬通過(guò)合成介孔非晶態(tài)載體制備還原型催化劑,用于愈創(chuàng)木酚和木質(zhì)素催化加氫,旨在為新能源的制備提供一種新方法。
1 實(shí)驗(yàn)部分
1.1 試劑與儀器
愈創(chuàng)木酚、庚烷、無(wú)水乙醇、硝酸銅、硝酸鈷、十六烷基三甲基溴化銨(CTAB,99%)、氫氧化鈉均為分析純;硫酸(98.3%)、偏鋁酸鈉均為化學(xué)純;十二烷,色譜純;硅溶膠(40%),工業(yè)品;商業(yè)ZSM載體(催化劑用),天津南開(kāi)催化劑廠;桉木屑木質(zhì)素,根據(jù)GB/T 2677.8—1994進(jìn)行提取。
Smart Lab 3型X射線衍射儀;NOVA2200e物理化學(xué)吸附儀;9790Ⅱ氣相色譜儀;7820A氣相-5977E質(zhì)譜氣相和質(zhì)譜聯(lián)用儀。
1.2 催化劑的制備
1.2.1 非晶態(tài)載體的制備 使用硅溶膠作為硅源,NaAlO2作為鋁源,CTAB作為模板劑,由NaOH、NaAlO2、硅溶膠、水和CTAB形成硅-鋁凝膠,凝膠各組分的摩爾比為n(SiO2)∶n(Al2O3)∶n(CTAB)∶n(Na2O)∶n(H2O)=20∶0.4∶1∶1.5∶400。
稱取一定量NaOH和NaAlO2,加水溶解。加入一定量的模板劑,混合、攪拌均勻,形成溶液A。將硅溶膠與適量的水混合,組成溶液B。在劇烈攪拌下,將溶液A逐滴加入B中,使用硫酸調(diào)節(jié)pH=10左右,得到凝膠,轉(zhuǎn)移至水熱反應(yīng)釜中,于180 ℃晶化48 h。過(guò)濾,用去離子水洗滌至洗液近中性,在105 ℃干燥12 h。在550 ℃馬弗爐中煅燒6 h,得到Na-型ZSM分子篩。
取一定量的Na-型分子篩,加入約10倍分子篩質(zhì)量的1 mol/L NH4NO3溶液于燒瓶中,在90 ℃油浴鍋中加熱攪拌5 h,重復(fù)3次。固體干燥后放于550 ℃馬弗爐中煅燒6 h,即得到氫型ZSM分子篩(ZS)。將氫型分子篩造粒、過(guò)篩,取40~80目固體作為分子篩載體,命名為ZS-S,S代表自制載體。
1.2.2 不同催化劑的制備 稱取一定量的硝酸鎳和硝酸鈷或硝酸銅或鉬酸銨,加水溶解。加入ZS-S載體,所得懸浮液放在搖床中,室溫下振蕩浸漬5 h。然后在80 ℃油浴鍋中將水分完全蒸干。稱取一定量的干燥樣品放置于石英管反應(yīng)器中,在400 ℃H2氛圍下還原4 h,氫氣流速為75 mL/min。催化劑中Ni∶M(金屬Co、Mo或Cu)的摩爾比為7∶3,活性金屬組分(NiO+MO)的總質(zhì)量為25%。根據(jù)助劑金屬種類的不同,分別命名為ZS-Cu、ZS-Mo和ZS-Co。
為了與商業(yè)ZSM載體相比,使用上述方法制備了助催化劑含Mo的Ni基催化劑,制備流程和金屬含量與上述相同,命名為ZSM-Mo。
1.3 催化加氫實(shí)驗(yàn)
催化加氫實(shí)驗(yàn)在體積為50 mL的間歇式高壓反應(yīng)釜中進(jìn)行。每次反應(yīng)加入2.0 g 愈創(chuàng)木酚或 0.5 g 木質(zhì)素、12 g的溶劑以及0.05 g催化劑,排空氣后填充一定壓力的H2,調(diào)節(jié)反應(yīng)溫度和轉(zhuǎn)速 (350 r/min),反應(yīng)至設(shè)定時(shí)間后,冷卻至室溫,收集液體產(chǎn)物,使用GC和GC-MS進(jìn)行分析。
1.4 液體產(chǎn)物分析
將收集的液體產(chǎn)物使用GC-MS和保留指數(shù)法進(jìn)行定性分析,使用GC-FID對(duì)其進(jìn)行定量分析。通過(guò)計(jì)算原料的轉(zhuǎn)化率(α)和產(chǎn)物選擇性(S)評(píng)價(jià)催化劑催化加氫活性。α和S的計(jì)算公式如下:
(1)
(2)
式中,ω1是液體產(chǎn)物中所有產(chǎn)物的含量;ω2是產(chǎn)物中原料的含量;ωx是液體產(chǎn)物中某一組分的含量。
2 結(jié)果與討論
2.1 催化劑的表征
2.1.1 XRD分析 圖1為載體ZS-S、催化劑ZS-Mo、ZS-Co、ZS-Cu和ZSM-Mo的XRD圖。
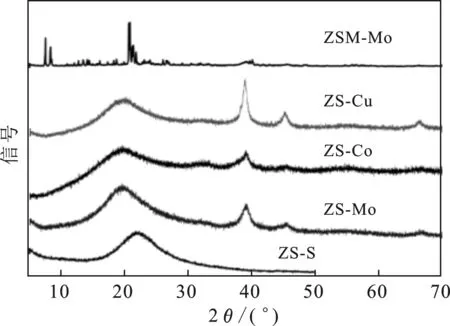
圖1 ZS-S、ZS-Mo、ZS-Co、ZS-Cu和ZSM-Mo的XRD分析
由圖1可知,ZS-S、ZS-Mo、ZS-Co、ZS-Cu四個(gè)樣品在20~30°出現(xiàn)了非晶態(tài)特征峰,說(shuō)明成功制備了非晶態(tài)載體和非晶態(tài)金屬催化劑。圖1中39.01,45.19,66.32°峰為金屬Ni單質(zhì)的特征峰,但三種非晶體催化劑中金屬Ni單質(zhì)峰具有顯著的差別,說(shuō)明不同助催化劑對(duì)金屬Ni的分散能力顯著不同。金屬Ni單質(zhì)衍射峰順序?yàn)椋篫S-Cu>ZS-Mo>ZS-Co,助劑Cu的加入最易引起金屬Ni單質(zhì)簇的形成。可能是因?yàn)镃u是三種金屬助劑中最易被還原產(chǎn)生金屬單質(zhì)的物質(zhì),銅單質(zhì)的形成可以為Ni2+的還原提供電子,致使金屬Ni組分容易被還原[8-9],同時(shí),非晶態(tài)載體酸性較弱(表1),對(duì)金屬組分的束縛能力較弱,有利于金屬Ni單質(zhì)的聚集。在非晶體催化劑中,助催化劑Co的加入,形成的Ni單質(zhì)峰最小,可能是由于金屬Co的加入提高了金屬Ni的分散性。樣品中未檢測(cè)到明顯的助催化劑衍射峰,可能是由于助催化劑金屬含量較低導(dǎo)致的[10-11]。與非晶態(tài)ZS-Mo催化劑相比,ZSM-Mo催化劑未出現(xiàn)非晶體特征峰,且金屬Ni單質(zhì)的衍射峰明顯較低,主要原因是晶體催化劑的酸性位點(diǎn)與金屬離子之間的作用力較強(qiáng),導(dǎo)致晶體催化劑中金屬組分的還原程度低,分散程度高[12-13]。
2.1.2 織構(gòu)性質(zhì)分析 表1為ZS-S載體和不同催化劑的織構(gòu)性質(zhì)。

表1 ZS-S、ZS-Mo、ZS-Co、ZS-Cu和ZSM-Mo的物化性質(zhì)
由表1可知,與非晶態(tài)ZS-S載體相比,ZS-Cu和ZS-Co的催化劑比表面積分別增加了124.2 m2/g和130.9 m2/g,而孔徑分別降低了1.97 nm和2.60 nm,催化劑中出現(xiàn)了微孔體積;比表面積的增加量和孔徑的降低程度均大于ZS-Mo。可能因?yàn)樨?fù)載和還原過(guò)程中形成的金屬微晶晶粒填充了ZS-S載體的部分介孔孔道,同時(shí)這些微晶晶粒也可能相互聚集,形成了新的微孔結(jié)構(gòu)[12]。助催化劑Co和Cu的離子半徑小于Mo,更易進(jìn)入載體孔道內(nèi)部,經(jīng)過(guò)還原形成的微晶晶粒更易阻塞孔徑,降低孔徑和增加比表面積。晶體ZSM-Mo催化劑微孔結(jié)構(gòu)發(fā)達(dá),雖然孔徑較小,但具有較大的比表面積。
2.1.3 NH3-TPD分析 ZS-Mo、ZS-Co、ZS-Cu和ZSM-Mo催化劑NH3-TPD圖見(jiàn)圖2。
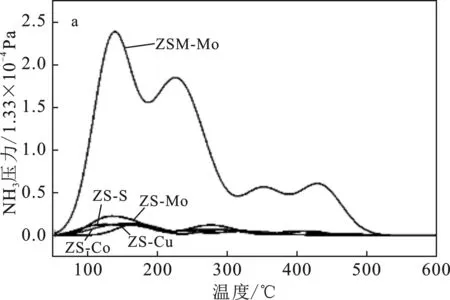
圖2 ZS-Mo、ZS-Co、ZS-Cu和ZSM-Mo催化劑NH3-TPD圖
由圖2a可知,晶體ZSM-Mo的峰顯著高于非晶體載體和催化劑,說(shuō)明晶體催化劑酸性較強(qiáng);非晶體ZS-S載體與非晶體催化劑脫附曲線也存在顯著的差異,表明金屬組分的負(fù)載對(duì)載體酸性具有一定的影響(見(jiàn)圖2b)。載體中原有的>400 ℃的強(qiáng)酸峰在負(fù)載金屬組分后,均明顯向低溫方向移動(dòng),而負(fù)載金屬后,200~400 ℃之間的峰強(qiáng)度有所增加,說(shuō)明金屬組分的負(fù)載一方面減少了載體表面的強(qiáng)酸性活性位點(diǎn),同時(shí)增加了中等酸性活性位點(diǎn)。NH3-TPD根據(jù)出峰溫度可以被劃分為弱酸峰(50~150 ℃)、中酸峰(150~400 ℃)和強(qiáng)酸峰(>400 ℃),各峰所占面積是通過(guò)高斯曲線模擬測(cè)量曲線進(jìn)行估算[13](表1)。
由表1可知,與非晶態(tài)ZS-S相比,在其上負(fù)載金屬組分并還原之后,催化劑酸量和強(qiáng)酸位點(diǎn)含量降低,催化劑中酸含量增加,說(shuō)明金屬組分易于和載體的強(qiáng)酸位點(diǎn)相結(jié)合,同時(shí)被還原的金屬組分可以為催化劑提供中酸活性位點(diǎn)。助催化劑Co的加入導(dǎo)致催化劑強(qiáng)酸位點(diǎn)的消失,可能是由于金屬Co最易與載體中的強(qiáng)酸性位點(diǎn)結(jié)合。晶體催化劑ZSM-Mo的酸量是非晶態(tài)催化劑ZS-Mo的3.5倍,且含有較多的強(qiáng)酸活性位點(diǎn)。
2.1.4 H2-TPR分析 圖3為ZS-Mo、ZS-Co和ZS-Cu的H2-TPR圖。
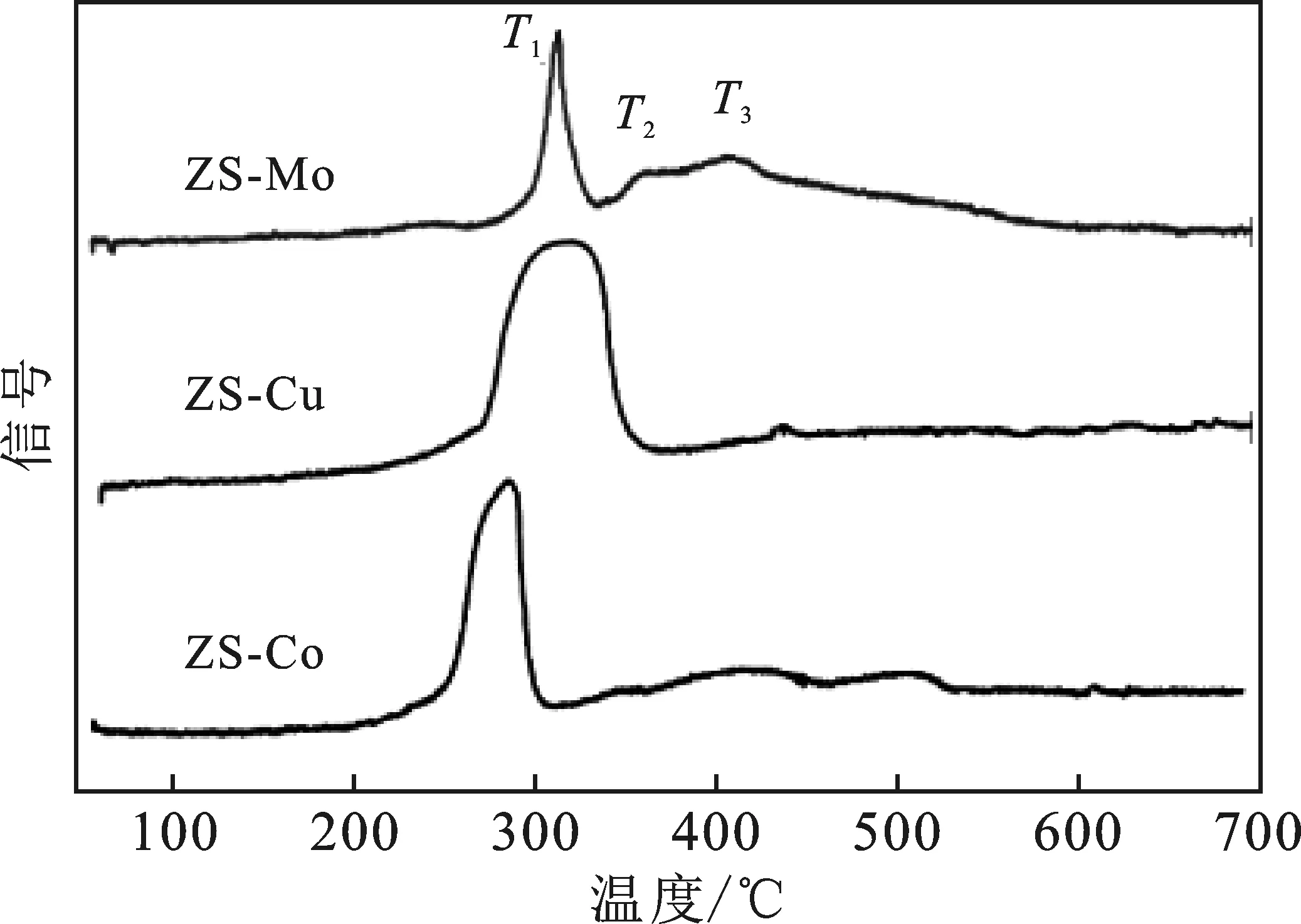
圖3 ZS-Cu、ZS-Co和ZS-Mo的H2-TPR圖
由圖3可知,對(duì)于ZS-Mo催化劑,在314 ℃ (T1)左右的峰屬于與載體結(jié)合力較弱的NiO的還原峰,在362 ℃(T2)處的峰歸屬于與載體結(jié)合力中等的金屬活性成分的還原峰,在408 ℃(T3)出現(xiàn)的峰歸因于非晶態(tài)聚合鉬酸鹽的還原峰[12]。由圖3可知,與ZS-Mo相比,ZS-Co的T1峰從314 ℃移到284 ℃,T2峰消失;ZS-Cu的T1和T2峰重合。結(jié)果表明,NiCo在較低的溫度下就開(kāi)始出現(xiàn)還原峰,說(shuō)明ZS-Co中Ni離子與載體結(jié)合較弱,具有較強(qiáng)的還原性;然而,在XRD分析中,ZS-Co催化劑的金屬Ni單質(zhì)的衍射峰較低,這可能是因?yàn)榻饘貱o的加入,促進(jìn)了Ni金屬原子在載體上的分散性而造成的。ZS-Cu中T1和T2峰重合,可能是由于金屬Cu的形成為屬Ni組分的還原提供了電子,降低了T2還原峰的溫度。
2.2 催化加氫活性
2.2.1 助催化劑對(duì)愈創(chuàng)木酚催化加氫活性的影響 不同助催化劑對(duì)催化加氫愈創(chuàng)木酚的產(chǎn)物組分、含量、原料轉(zhuǎn)化率和產(chǎn)物選擇性見(jiàn)表2。反應(yīng)條件:溫度180 ℃,4 MPa H2,轉(zhuǎn)速為 350 r/min,乙醇為溶劑。烷烴的選擇性可以反映加氫脫氧反應(yīng)的程度;脂肪族含氧衍生物的選擇性可以反映苯環(huán)的加氫程度;芳香族含氧衍生物的含量可以反映愈創(chuàng)木酚中甲氧基和酚羥基加氫程度[14]。

表2 不同催化劑催化愈創(chuàng)木酚加氫的產(chǎn)物組成和選擇性
由表2可知,ZS-Co對(duì)愈創(chuàng)木酚的催化加氫產(chǎn)物中環(huán)己酮、環(huán)己醇和1-甲基-1,2-環(huán)己二醇等成分的含量遠(yuǎn)高于ZS-Mo和ZS-Cu,而且原料的轉(zhuǎn)化率最高,芳香族含氧衍生物的選擇性最低,主要是由于金屬Co的引入增強(qiáng)了催化劑對(duì)C—O鍵的氫解能力,同時(shí)促進(jìn)了金屬Ni活性位點(diǎn)的分散性[14-15],因此,有利于愈創(chuàng)木酚中甲氧基C—O和苯環(huán)的深度加氫反應(yīng)。ZS-Cu和ZSM-Mo催化劑雖然轉(zhuǎn)化率較低,但是產(chǎn)物中烷烴的選擇性較高,表明催化劑雖然對(duì)愈創(chuàng)木酚的催化活性較低,但是產(chǎn)物的加氫程度較高,ZS-Cu產(chǎn)生的原因可能是由于Cu加入,使金屬Ni活性位點(diǎn)大量的聚集,有利于活化產(chǎn)生H原子[14];ZSM-Mo催化劑產(chǎn)生的原因可能是催化劑酸性較強(qiáng),對(duì)含氧化合物的吸附能力強(qiáng),中間產(chǎn)物未完全脫氧,不能從催化劑的表面脫除,但中間產(chǎn)物會(huì)聚合產(chǎn)生積碳,造成催化劑活性降低。
2.2.2 溶劑對(duì)愈創(chuàng)木酚催化加氫產(chǎn)物的影響 不同助催化劑對(duì)愈創(chuàng)木酚的催化加氫活性有較大的影響,但在上述反應(yīng)條件下,原料的轉(zhuǎn)化率較低,這可能是因?yàn)橐掖贾械臉O性官能團(tuán) —OH基與反應(yīng)原料分子中的氧元素在催化劑表面發(fā)生競(jìng)爭(zhēng)吸附行為,乙醇中的羥基占據(jù)了酸性和金屬活性位點(diǎn),阻礙了原料的吸附、活化和加氫反應(yīng)。為了對(duì)比乙醇對(duì)加氫產(chǎn)物的影響,本文使用非極性溶劑-庚烷做對(duì)照實(shí)驗(yàn)。反應(yīng)條件:溫度180 ℃,4 MPa H2,轉(zhuǎn)速為 350 r/min,正庚烷為溶劑,結(jié)果見(jiàn)表3。

表3 在庚烷溶劑中愈創(chuàng)木酚催化加氫的產(chǎn)物組成和選擇性

2.2.3 木質(zhì)素加氫產(chǎn)物的研究 介孔Ni基催化劑ZS-Mo和ZSM-Mo對(duì)桉木屑木質(zhì)素催化加氫產(chǎn)物的組成和含量見(jiàn)表4。實(shí)驗(yàn)條件:溫度350 ℃,4 MPa H2,反應(yīng)3 h,水為溶劑。
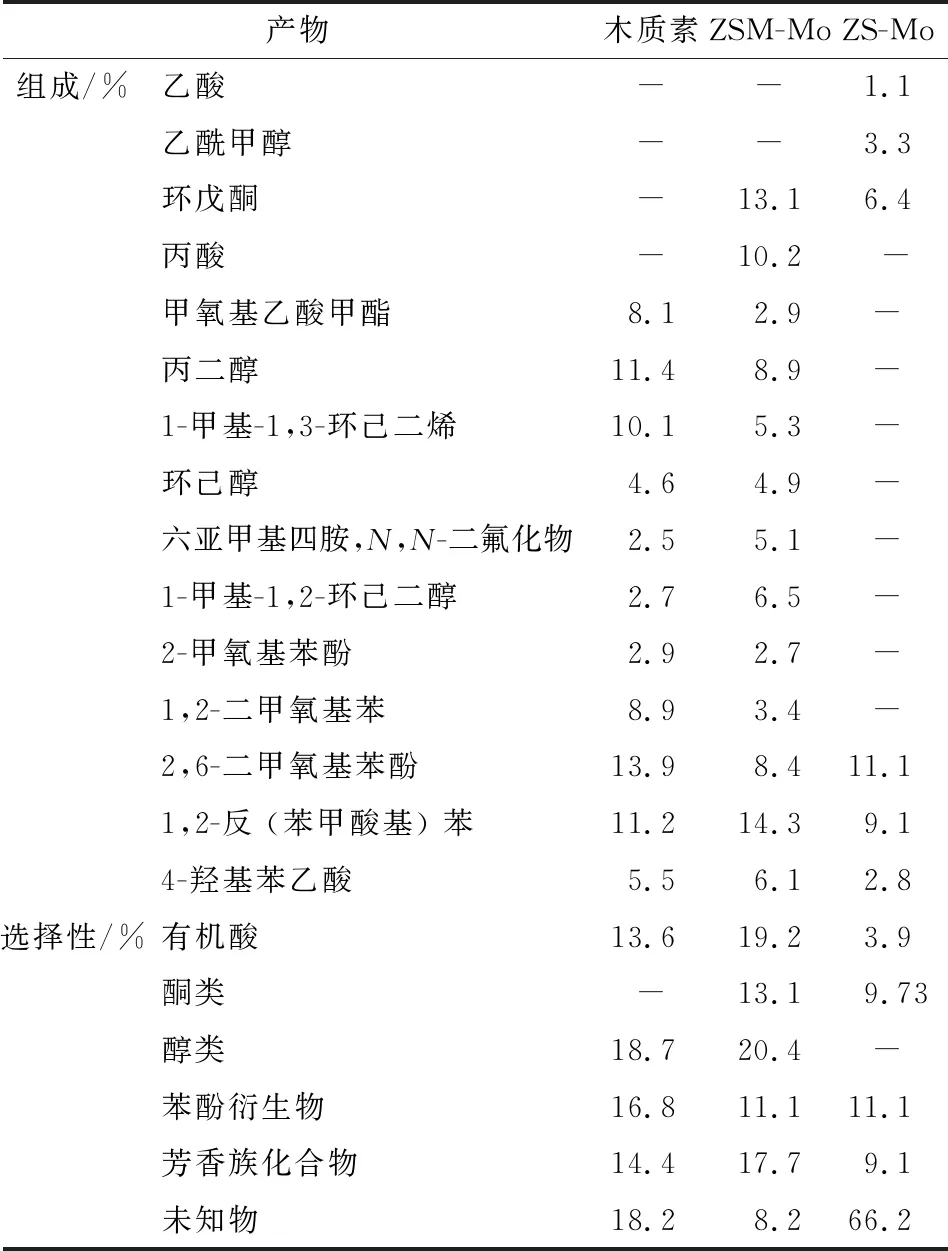
表4 ZSM-Mo和ZS-Mo催化精制木質(zhì)素的主要產(chǎn)物組成和含量與木質(zhì)素水溶性主要組成
由表4可知,木質(zhì)素原料水溶性化學(xué)組成主要包括有機(jī)酸、醇類和酚類等物質(zhì)。經(jīng)過(guò)ZSM-Mo催化加氫后,產(chǎn)物中水溶性組分增加,同時(shí)出現(xiàn)了酮類產(chǎn)物。ZSM-Mo催化劑酸性較強(qiáng),有助于木質(zhì)素的水解,同時(shí)金屬活性位點(diǎn)對(duì)降解產(chǎn)物進(jìn)一步催化加氫,導(dǎo)致酮類物質(zhì)選擇性增加。而孔徑較大酸性較弱的ZS-Mo催化劑,產(chǎn)物中水溶性組分減少,尤其是醇類的選擇性顯著降低,可能是由于非晶態(tài)催化劑對(duì)水解產(chǎn)物具有較強(qiáng)的催化加氫能力;但未知物的選擇性增加,酸性較弱的催化劑不利于木質(zhì)素的降解反應(yīng),甚至由于吸附能力較弱,中間產(chǎn)物從催化劑表面脫離而相互發(fā)生聚合反應(yīng)。
2.2.4 木質(zhì)素加氫精制前后的對(duì)比 圖4為木質(zhì)素水溶液加氫精制反應(yīng)前后的對(duì)比。

圖4 木質(zhì)素加氫前后的對(duì)比
由圖4可知,桉木屑木質(zhì)素部分溶于水,反應(yīng)前木質(zhì)素的水溶液中可以觀察到明顯的木質(zhì)素固體顆粒;經(jīng)過(guò)加氫精制之后,溶液由棕色濁液變?yōu)樽厣该饕后w,產(chǎn)品溶液中的固體懸浮顆粒含量顯著降低,表明木質(zhì)素得到了一定程度的降解。
3 結(jié)論
制備了非晶態(tài)還原型Ni基催化劑,分別對(duì)愈創(chuàng)木酚和木質(zhì)素進(jìn)行了催化加氫反應(yīng),研究了助催化劑種類和溶劑對(duì)催化愈創(chuàng)木酚加氫產(chǎn)物的影響,及非晶態(tài)催化劑對(duì)催化加氫木質(zhì)素產(chǎn)物的影響。主要結(jié)果如下:
(1)不同金屬助催化劑對(duì)催化劑的性質(zhì)有顯著的影響。助催化劑Co有利于還原型Ni基催化劑上Ni活性位點(diǎn)的分散。
(2)在愈創(chuàng)木酚的催化加氫實(shí)驗(yàn)中,金屬Co的引入,有利于愈創(chuàng)木酚催化活性的增加;Cu的引入和酸性的增加有利于烷烴的生成。非極性溶劑有利于愈創(chuàng)木酚的催化轉(zhuǎn)化,催化反應(yīng)路徑未發(fā)生明顯變化。
(3)木質(zhì)素在非晶體還原性Ni基催化劑作用下,降解產(chǎn)物發(fā)生了進(jìn)一步加氫反應(yīng)。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時(shí)代(2018年3期)2018-06-11 16:10:44
浙江大學(xué)學(xué)報(bào)(工學(xué)版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國(guó)資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(xué)(2015年4期)2016-01-17 09:01:27
應(yīng)用化工(2014年3期)2014-08-16 13:23:50
