有機-無機磷鎢酸鹽無溶劑催化氧化環己烯制己二酸
2021-06-15 07:31:18王璐璐曾祥瑞史慶偉王海彥王鈺佳
石油化工 2021年5期
關鍵詞:催化劑
王璐璐,曾祥瑞,史慶偉,王海彥,王鈺佳
(1. 中國石油大學(華東) 化學工程學院,山東 青島 266580;2. 遼寧石油化工大學 石油化工學院,遼寧 撫順 113001)
己二酸是合成尼龍-66和化工塑料的重要原料[1-2],工業上主要采用硝酸氧化法或環己烷兩步氧化法合成[3]。上述方法的己二酸收率和選擇性雖然較高,但是面臨生產設備腐蝕較嚴重、硝酸氧化排放N2O嚴重破壞生態環境等問題[4-5]。近年來,催化劑聯合H2O2直接氧化環己烯制己二酸工藝由于具有工藝路線短、清潔環保和投資成本低等優點而備受關注。Sato等[6]首次開發了以Na2WO4為催化劑、H2O2為氧化劑,直接催化氧化環己烯制己二酸的清潔生產工藝,為環己烯清潔催化制備己二酸的研究提供了以鎢元素為活性中心的思路。隨后,各種基于鎢酸鹽的均相催化劑相繼得到了研究,如(H2en)3[P2W18O62]·6.48H2O[7]、WO(O2)2·2QOH[8]和H3PW12O40[9]等。這些研究結果表明,鎢酸鹽具有顯著的環己烯氧化制己二酸的催化活性。但這些均相催化劑良好的水溶性導致了反應后產物難以提純,催化劑回收困難等問題。為此,一些雜多酸鹽,如Cs3PW12O40[10]、K3PW12O40[1]等非均相催化劑被開發出來以緩解上述問題,但在制備己二酸過程中添加的大量溶劑與產物之間的分離又成為新的問題。在多金屬氧酸鹽中添加醋酸[8]、鹽酸[11]、酸性離子液體[12]等B酸助劑有助于提高己二酸收率,但該工藝存在離子液體或酸性溶劑浸出等問題。近年來,基于雜多酸的有機-無機雜化材料的合成及應用得到了廣泛關注,該類材料與單一的雜多酸相比,具有催化活性高、助劑依賴性低等優點。
本工作以1-(3-磺丙基)嗎啡啉為有機陽離子、磷鎢酸(HPW)為無機陰離子,制備了有機-無機雜多酸鹽,利用FTIR,TG,XRF,GC-MS,UVVis等方法考察了雜多酸鹽的性能及催化環己烯氧化制己二酸的活性,并探討了催化反應機理。
1 實驗部分
1.1 主要試劑與儀器
N-甲基嗎啡啉、HPW、環己烯、H2O2(30%(w))、N,N-二甲基甲酰胺(DMF)、無水乙醇、丙酮:分析純,國藥集團化學試劑有限公司;1,3-丙烷磺酸內酯:純度99%(w),武漢中德遠東精細化工有限公司;實驗用水均為去離子水。
Spectrum One型傅里葉變換紅外光譜儀:Perkin Elmer公司;Q600型熱重-差熱分析儀:TA公司;Agilent Cary 5000型紫外可見近紅外分光光度計、Agilent 7890A/5975C型氣質聯用儀:安捷倫科技有限公司;S8 Tiger波長型X射線熒光光譜儀:Bruker AXS公司。
1.2 催化劑制備
將一定量1,3-丙烷磺酸內酯溶于丙酮中,在冰浴下用滴液漏斗向反應器中邊攪拌邊滴加N-甲基嗎啡啉(1,3-丙烷磺酸內酯與N-甲基嗎啡啉的摩爾比為1∶1),反應1 h后生成的白色固體用丙酮洗滌3次,80 ℃下干燥后得中間體1-(3-磺丙基)嗎啡啉。
取一定量脫除結晶水后的HPW溶于去離子水中,然后加入1-(3-磺丙基)嗎啡啉(1-(3-磺丙基)嗎啡啉與HPW的摩爾比為3∶1),室溫下反應24 h,將所得產物減壓蒸餾脫水,將得到的固體置于真空干燥箱中80 ℃下干燥24 h,得灰綠色固體,即為1-(3-磺丙基)嗎啡啉磷鎢酸鹽([C3SO3Hnhm]3PW12O40)。
N-甲基-N-丁基嗎啡啉磷鎢酸鹽([Nbmm]3·PW12O40)參考本課題組前期工作[13]合成。
1.3 催化氧化反應
催化氧化環己烯制己二酸的反應在裝有溫度計和恒壓滴液漏斗的三口燒瓶(100 mL)中進行。先將一定量催化劑與H2O2均勻混合,然后打開恒壓滴液漏斗旋塞,緩慢滴加環己烯(50 mmol,5 mL),攪拌至反應設定時間后停止加熱,冰水浴冷卻、離心分離,將液體減壓蒸餾,得到固體催化劑,剩余液體用GC-MS方法分析組成。將離心分離折出的白色固體用冷水洗滌后重結晶,再抽濾、烘干得到純白色晶體即為己二酸,它的FTIR譜圖和熔點(151~153 ℃)與標準試樣一致。產物己二酸收率(Y)的計算見式(1)。

式中,m1為實際得到的己二酸晶體質量,g;m2為按照化學計量式計算的理論己二酸晶體質量,g。
1.4 催化劑結構表征
采用KBr壓片法進行FTIR測試,掃描波數范圍400~4 000 cm-1。
TG測試:氮氣氛圍,升溫速率10 ℃/min,室溫~700 ℃。
XRF測試:管功率4 kW,Rh靶,75 mm鈹窗,采用硼酸包邊固體制樣法制樣,best測試模式。
GC-MS測試:HP-5MS柱(30 m×250 μm×0.25 μm),進樣口溫度280 ℃,50 ℃保持2 min,然后以10 ℃/min升溫至300 ℃保持5 min。
UV-Vis測試:測試試樣在乙腈溶液中的吸光度,波長范圍200~800 nm。
試樣的酸強度測定方法[12]:以對硝基苯胺為指示劑,配制對硝基苯胺-乙腈溶液,待測試樣配制成不同濃度的乙腈溶液。向待測試樣中加入一定量對硝基苯胺-乙腈溶液,利用UV-Vis法測定上述溶液在最大吸收波長下的吸光度,由式(2)計算試樣的Hammett酸函數(H0)。

式中,pKa為指示劑的解離常數,對硝基苯胺的pKa為0.99;xB為指示劑未被質子化部分的摩爾分數,%;xBH+為指示劑在溶劑中的質子化部分的摩爾分數,%。
2 結果與討論
2.1 UV-Vis表征結果
鎢系雜多化合物在紫外可見區有很強的吸收,通過UV-Vis表征,能夠觀察到過渡金屬的d-d躍遷以及由電荷轉移引起的其他躍遷。試樣的UV-Vis譜圖見圖1。從圖1可看出,試樣均在200~400 nm范圍內有吸收峰。其中在206 nm附近是O→P的吸收峰,而在265 nm附近出現的吸收峰是O2-W6+之間的金屬電荷遷移[14]。在[C3SO3Hnhm]3PW12O40中206 nm處的吸收峰發生了藍移,這表明離子液體和HPW陰離子雜化材料之間存在一定的相互作用。
試樣在乙腈溶液中的H0見表1。

圖1 試樣的UV-Vis譜圖Fig.1 UV-Vis spectra of the samples.
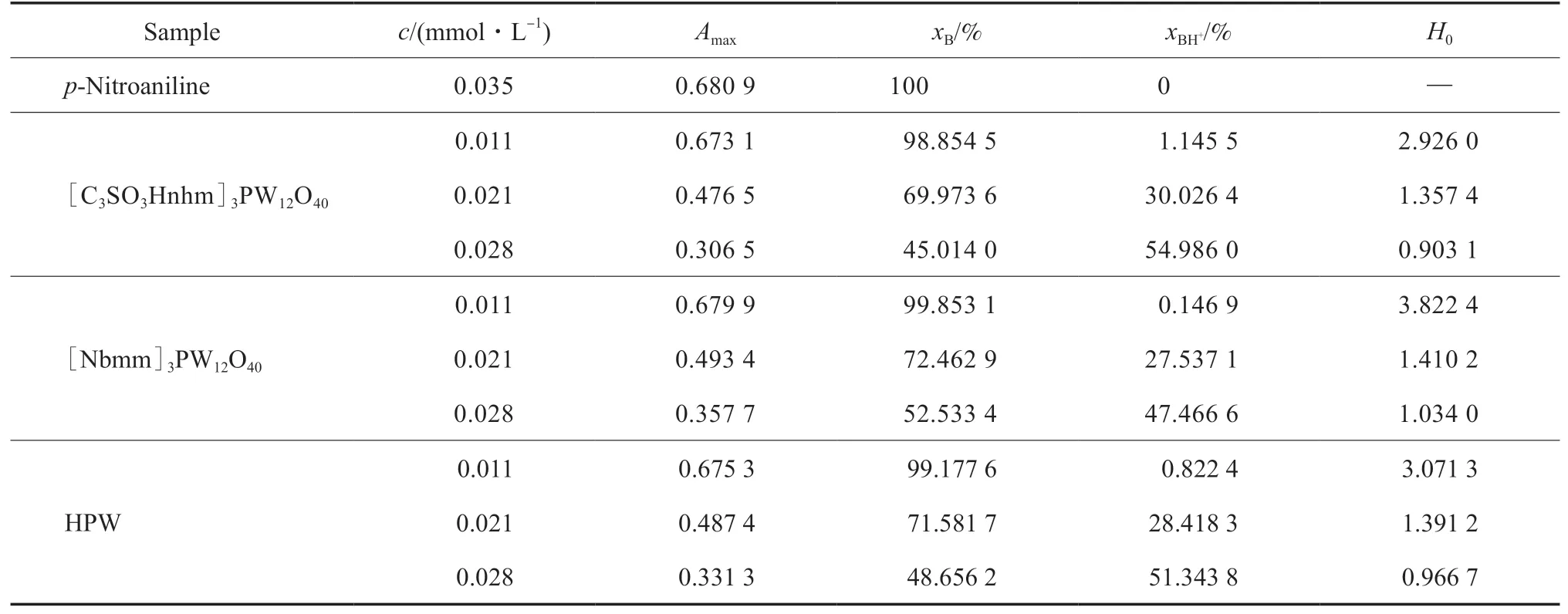
表1 試樣的H0Table 1 Hammett-function(H0) of the samples
從表1可看出,隨濃度的增大,試樣的H0均減小;相同濃度下,不同試樣的H0由大到小的順序為:[Nbmm]3PW12O40>HPW>[C3SO3Hnhm]3PW12O40,說明在乙腈溶液中,相同濃度下[C3SO3Hnhm]3PW12O40的酸強度最大。
2.2 XRF表征結果
試樣的XRF表征結果見表2。從表2可以看出,各元素含量的測試值與理論值基本相符,說明制備的試樣與預期結構一致。[C3SO3Hnhm]3PW12O40重復催化使用6次后的元素含量與新鮮催化劑相比基本沒有改變,說明[C3SO3Hnhm]3PW12O40具有較好的重復使用性。
2.3 催化劑的熱穩定性
試樣的TG曲線見圖2。從圖2可以看出,[C3SO3Hnhm]3PW12O40和[Nbmm]3PW12O40的熱分解溫度分別為250,380 ℃,這可能是由于磺酸基團的引入減弱了分子間作用力;升溫至650 ℃后,二者的失重曲線基本穩定,且失重率分別達到18.36%和18.91%,說明它們的有機陽離子已基本分解完全。

表2 試樣的XRF表征結果Table 2 XRF results of the samples

圖2 試樣的TG曲線Fig.2 TG curves of the samples.
2.4 催化劑的活性
不同催化劑氧化環己烯制己二酸的催化活性見圖3。

圖3 催化劑的催化活性Fig.3 Catalytic activity of catalysts.
從圖3可看出,三種催化劑的活性大小順序為:[C3SO3Hnhm]3PW12O40>HPW>[Nbmm]3PW12O40,這與H0表示的酸強度結果一致,說明催化劑酸性對催化活性有一定影響。[C3SO3Hnhm]3PW12O40由于不含較強的自由基激發活性位點,保證了反應體系不會沿自由基路線反應,而是沿雙鍵氧化路線進行[12,15],含有的磺酸基酸性位點能夠活化水分子,并與中間產物環氧環己烷反應生成1,2-環己二醇,再進一步反應生成己二酸,這與王新國等[16]的研究結果基本一致。
2.5 反應條件的影響
反應條件對己二酸收率的影響見圖4。從圖4a可看出,隨反應時間的延長,己二酸收率增大,當反應時間達10 h時,己二酸收率可達85.3%;此后繼續延長反應時間,己二酸收率變化不大。因此,選擇10 h為最佳反應時間。從圖4b可看出,隨溫度的升高,反應速率加快,己二酸收率增大,但溫度超過90 ℃后,己二酸收率增幅減小。這可能是因為,H2O2氧化環己烯需要更高的反應溫度才能實現氧原子的轉移[17],但溫度過高,H2O2均裂生成羥基自由基,會引發烯丙基副反應[18]。為了保證反應體系沿雙鍵氧化的路線反應,反應體系的溫度不宜過高,因此,最佳的反應溫度為90 ℃。從圖4c可看出,催化劑用量的增大可大幅提高己二酸收率,當催化劑加入量達到1.0 g時,己二酸收率達到最高;繼續增加催化劑加入量,己二酸收率反而下降,可能是因為過量的催化劑反而加快了副反應的發生。因此,最佳的催化劑加入量為1.0 g。由圖4d可看出,隨H2O2用量的增加,己二酸收率逐漸提高,當n(H2O2)∶n(環己烯)=4.4時,己二酸收率達到最大,此后繼續增加H2O2用量,己二酸收率反而降低,可能是因為過量的H2O2導致了副反應加劇。因此,最佳的n(H2O2)∶n(環己烯)=4.4。
2.6 催化劑的重復使用性能
為了考察[C3SO3Hnhm]3PW12O40的重復使用效果,反應結束后將分離得到的固體催化劑用溶劑DMF洗滌3次,然后在烘箱中150 ℃下干燥至恒重,重復用于環己烯氧化反應,結果見圖5。

圖4 工藝條件對己二酸收率的影響Fig.4 The effect of reaction conditions on the yield of AA.
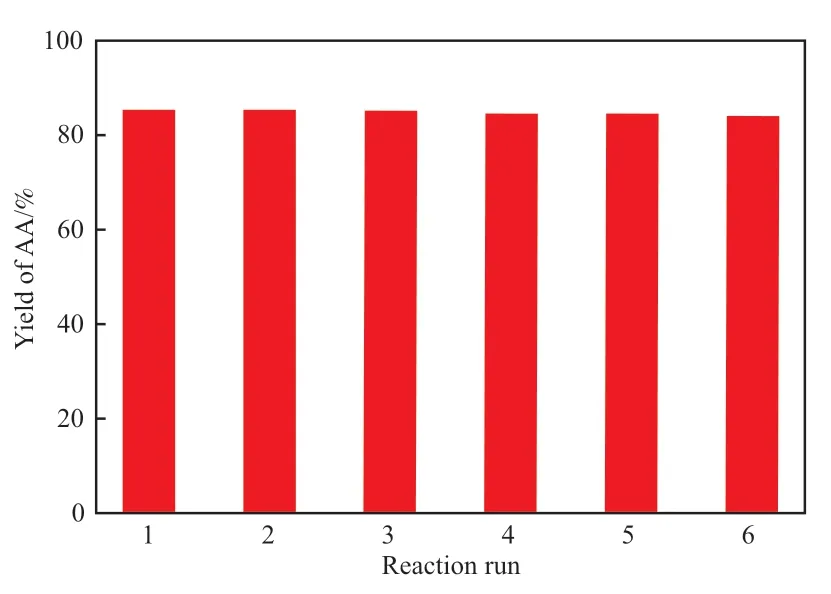
圖5 [C3SO3Hnhm]3PW12O40重復使用性能Fig.5 The reusability of[C3SO3Hnhm]3PW12O40.Reaction conditions referred to Fig.3.
從圖5可以看出,[C3SO3Hnhm]3PW12O40重復使用6次后,己二酸收率仍能達到84%。
[C3SO3Hnhm]3PW12O40使用前和重復使用6次后的FTIR譜圖見圖6。由圖6可知,兩個試樣均存在屬于Keggin結構的4個特征吸收峰[19],分別為1 078(P-Oa),982(W-Od),894(W-Ob-W),803(W-Oc-W) cm-1。3 442 cm-1(O—H鍵伸縮振動),2 967 cm-1和2 873 cm-1(甲基中C—H鍵的不對稱和對稱伸縮振動),1 470 cm-1(—CH3),1 633 cm-1(季銨基團),1 118 cm-1(C—N鍵伸縮振動),1 165 cm-1(—S=O)均屬于有機陽離子的特征峰。FTIR表征結果顯示,[C3SO3Hnhm]3PW12O40使用前后均保留了有機陽離子和雜多陰離子的結構。

圖6 [C3SO3Hnhm]3PW12O40使用前(a)和重復使用6次后(b)的FTIR譜圖Fig.6 FTIR spectra of [C3SO3Hnhm]3PW12O40 before(a) and after used for 6 times(b).
2.7 催化反應機理
有機-無機磷鎢酸鹽催化氧化環己烯制己二酸反應發生在催化劑表面。為了探究反應機理,在反應9 h后,對析出己二酸晶體后剩余的液體進行GC-MS分析,結果見圖7。從圖7可看出,除了主產物己二酸外,還生成了1,2-環己二醇等,這些都是環己烯雙鍵氧化的中間產物,初步估計環己烯在[C3SO3Hnhm]3PW12O40的作用下是沿著雙鍵氧化的路徑進行反應。反應機理見圖8。
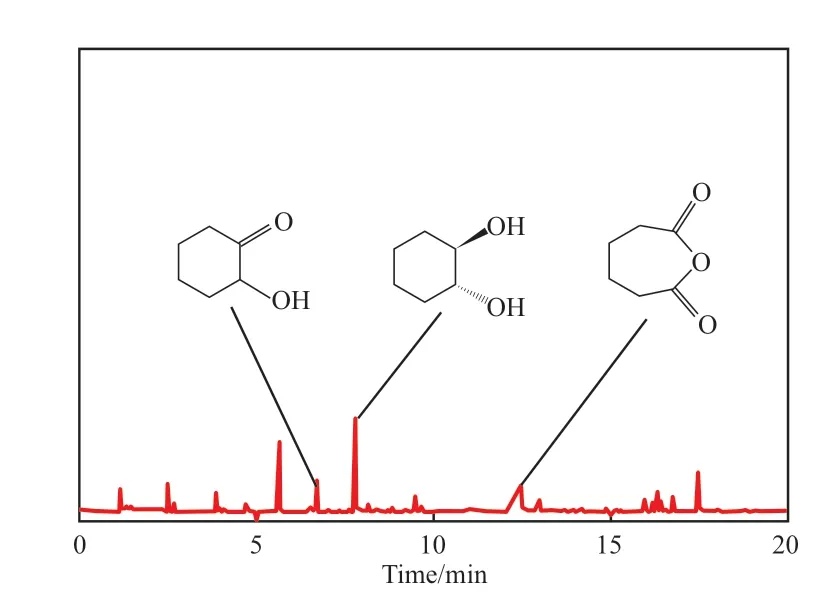
圖7 液體的GC-MS譜圖Fig.7 GC-MS spectra of liquid.

圖8 反應機理Fig.8 The reaction mechanism.
從圖8可看出,W首先被H2O2氧化為過氧鎢酸鹽[WO(O2)2(OH)2]2-,[WO(O2)2(OH)2]2-是環己烯氧化的催化活性物質,環己烯雙鍵氧化生成環氧環己烷;有機配體上的磺酸提供了B酸,在B酸作用下環氧環己烷發生水解進一步被氧化為1,2-環己二醇,最終被氧化為己二酸。由于[C3SO3Hnhm]3PW12O40不含較強的自由基激發活性位點,因而在催化環己烯氧化制己二酸的反應中表現出較強的催化性能。而[WO(O2)2(OH)2]2-被還原為PW12O3-40,催化劑得以重復使用,H2O2被不斷消耗。
2.8 催化氧化對比實驗
[C3SO3Hnhm]3PW12O40與其他催化劑進行對比的結果見表3。由表3可知,本工作最優工藝條件下的己二酸收率為85.3%,遠高于文獻中[CH3(CH2)15N(CH3)3]3{PO4[WO(O2)2]4},K3PW12O40,Na2WO4·(H2O)2的己二酸收率。盡管H2WO4的己二酸收率有96.0%,但它需要在離子液體中反應,而離子液體制備成本高,且回收過程存在分離和流失問題。因此,和其他固體催化體系相比,[C3SO3Hnhm]3PW12O40展現了更高的催化活性和更好的經濟適應性。

表3 [C3SO3]3PW12O40和其他固體催化劑的催化性能對比Table 3 Comparison of catalytic performance between[C3SO3Hnhm]3PW12O40 and reported solid catalysts
3 結論
1)制備了有機-無機磷鎢酸鹽[C3SO3Hnhm]3·PW12O40和[Nbmm]3PW12O40,酸強度由大至小順序為[C3SO3Hnhm]3PW12O40>HPW>[Nbmm]3PW12O40,與催化氧化環己烯制備己二酸的催化活性一致。
2)在反應溫度90 ℃、反應時間10 h、催化劑加入量1.0 g、H2O2用量22 mL、環己烯用量5 mL(n(H2O2)∶n(環己烯)=4.4)的條件下,[C3SO3Hnhm]3PW12O40表現出最佳的催化活性,己二酸收率達到85.3%。催化劑循環使用6次后結構仍沒有變化,且己二酸收率沒有明顯下降。與其他催化劑對比,[C3SO3Hnhm]3PW12O40顯示了更高的催化活性和更好的經濟適應性。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
