淫羊藿不同部位有效成分含量比較研究
2021-06-16 01:44:38鄧寒霜楊文怡
陜西農業科學 2021年4期
鄧寒霜,楊文怡,賀 博,李 月
(1.商洛學院 生物醫藥與食品工程學院,陜西 商洛 726000;2.陜西秦嶺特色生物資源產業技術研究院,陜西 商洛 726000;3.陜西天士力植物藥業有限公司,陜西 商洛 726000)
中藥淫羊藿具有悠久的應用歷史,具有補肝腎、袪風濕、強筋骨等功效,在我國最早的藥學專著《神農本草經》中就有收錄,列為上品[1~3]。我國是淫羊藿屬植物地理分布中心,分布有40余種淫羊藿,2005年版《中華人民共和國藥典》(以下簡稱"藥典")收載淫羊藿藥材的來源有5種小檗科植物,分別是淫羊藿(EpimediumbrevicornuMaxim)、朝鮮淫羊藿(EpimediumkoreanumNakai)、箭葉淫羊藿(Epimediumsagittatum(Sieb.et Zucc) Maxim)、柔毛淫羊藿(EpimediumpubescensMaxim)以及巫山淫羊藿(EpimediumwushanenseT. S. Ying)的干燥地上部分[4]。從2010年起,巫山淫羊藿因為與其他幾種來源的淫羊藿藥材在化學成分上有較大的區別,而被單獨作為一味中藥收錄于藥典中。自此,藥典關于淫羊藿藥材的植物來源開始減少成4種,分別是淫羊藿、箭葉淫羊藿、柔毛淫羊藿、朝鮮淫羊藿;同時,將淫羊藿藥用部位由"干燥地上部分"變更為"干燥葉"[5]。至目前最新發行的2020年版藥典,淫羊藿藥材來源一直沿用2010年的相關規定[6,7]。為驗證淫羊藿藥用部位劃分的合理性,比較淫羊藿植物不同部位中藥用成分含量的差異,以期為擴大淫羊藿藥材來源,減少自然資源的浪費提供依據,本文對來自淫羊藿不同部位的樣品材料中幾種有效成分的含量進行了對比分析。
1 材料與方法
1.1 材料與設備
淫羊藿原植物,采集自陜西、甘肅等地,由商洛學院執業藥師李筱鑒定為小檗科植物淫羊藿(EpimediumbrevicornuMaxim)或柔毛淫羊藿(EpimediumpubescensMaxim),樣品來源詳細信息見表1。淫羊藿苷對照品(批號201516),購自中國藥品生物制品檢驗所;朝藿定A(批號1804027)、朝藿定B(批號19071403)、朝藿定C(批號190625)、寶藿苷Ⅰ(批號1804026)等對照品購自北京世紀奧科生物技術有限公司。
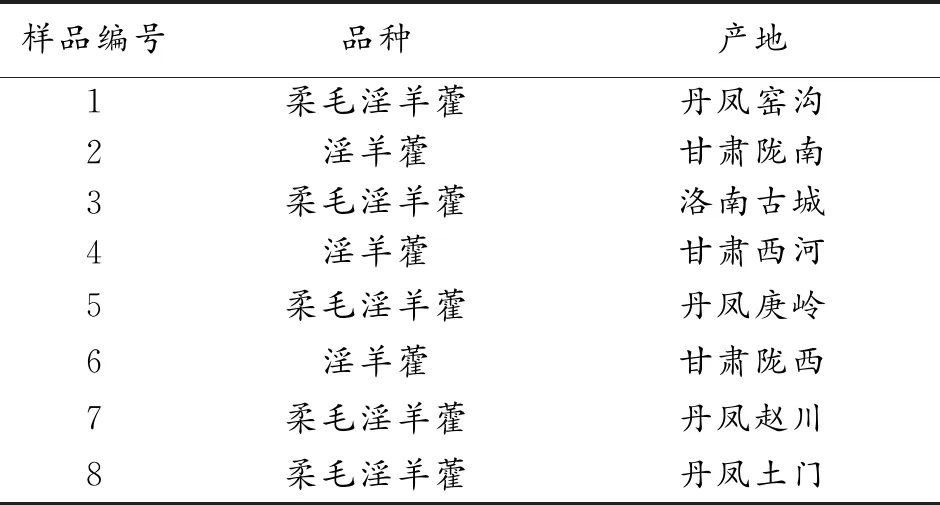
表1 淫羊藿樣品產地信息
檢測使用的儀器為日本島津公司出品的LC-20A型HPLC儀,配有在線脫氣機、高壓泵、四元低壓梯度混合裝置、二極管陣列檢測器、原裝自動進樣器、原裝柱溫箱、島津LabSolutions色譜工作站等。
1.2 方法
1.2.1 樣品處理 將淫羊藿原植物除去雜質,按根、莖、葉柄、葉片等不同部位分成4類供試品,干燥后粉碎,過3號藥典篩(24目,篩孔內徑355 μm±13 μm),即得供試品粉末。
1.2.2 含量測定 色譜條件 采用十八烷基鍵合的硅膠色譜柱(250 mm×4.6 mm,填料粒度5 μm),以乙腈(A)、水(B)為流動相,按表2中規定進行梯度洗脫,檢測波長270 nm,柱溫30 ℃,進樣量10 μL,記錄60 min內色譜流出曲線。
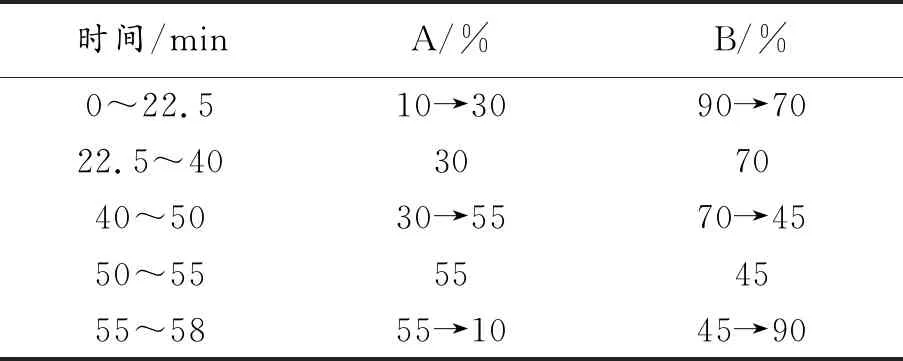
表2 淫羊藿樣品含量測定梯度洗脫順序
供試品溶液的制備 取粉碎后通過三號藥典篩的樣品0.2 g左右,用萬分之一的電子天平準確稱量樣品的質量,將樣品粉末裝于具塞磨口瓶中,加入20 mL稀乙醇作為提取溶劑后,立即塞密,稱取并記錄其重量,采用超聲波提取法提取樣品(功率400 W,頻率50 kHz),提取時間為1 h 。提取完成后,取出樣品溶液放冷,再次帶塞稱取瓶中提取溶液的重量與前述記錄比較,補充損失的稀乙醇,將樣品提取溶液搖勻后,用微孔濾膜過濾,取續濾液,即得供試品溶液[7-9]。
對照品溶液的制備 取朝藿定A、B、C,寶藿苷Ⅰ以及淫羊藿苷等5種待測組分的對照品適量,準確稱取其質量,用甲醇作為溶劑溶解并轉移至體積為25 mL的容量瓶內,再以甲醇為溶劑定容至刻度,即得對照品溶液。
2 結果與分析
2.1 系統適應性考察結果
2.1.1 精密度考察結果 準確量取上述對照品的溶液10 μL注入HPLC儀進行分析,記錄各組分色譜峰的峰面積,連續進樣5次以上,經計算后得到淫羊藿苷,朝藿定A、B、C和寶藿苷Ⅰ 等5個組分各自色譜峰峰面積的RSD值分別為0.21%、0.32%、0.24%、0.31%、0.35%,考察結果證明了儀器具有良好的精密度。
2.1.2 重復性考察結果 利用同一個淫羊藿藥材的樣品粉末,依據前述的方法處理樣品成為其供試品溶液,重復處理樣品溶液5份以上,準確量取該供試品溶液10 μL注入HPLC儀進行分析,記錄各組分色譜峰的峰面積,經計算后得到淫羊藿苷,朝藿定A、B、C和寶藿苷Ⅰ 等5個組分各自色譜峰峰面積的RSD值分別為0.83%、0.87%、0.74%、0.86%、1.01%,表明研究所用供試品制備方法具有良好的重復性。
2.1.3 穩定性考察結果 取同一供試品溶液,照1.2.2項中色譜條件,分別間隔0 h、4 h、8 h、12 h、24 h后,準確量取該供試品溶液10 μL注入HPLC儀進行分析,記錄各組分色譜峰的峰面積,經計算后得到淫羊藿苷,朝藿定A、B、C和寶藿苷Ⅰ 等5個組分各自色譜峰峰面積的RSD值分別為2.03%、3.46%、2.81%、3.27%、3.79%,穩定性考察試驗結果證明供試品的溶液其內在品質在24 h內不會發生較大變化。
2.1.4 校準曲線繪制 分別吸取淫羊藿苷、朝藿定A、朝藿定B、朝藿定C、寶藿苷Ⅰ 不同濃度的對照品溶液進樣分析,記錄色譜數據,以各有效成分的濃度為橫坐標(X),以峰面積為縱坐標(Y),繪制校準曲線,得各對照品曲線方程如下。
淫羊藿苷:Y=22 990 302.7778X-35 033.3333,R2=0.9999,線性范圍0~0.36 mg·mL-1。
朝藿定A:Y=17 058 760.7143X+3 577.9524,R2=0.9996,線性范圍0~0.32 mg·mL-1。
朝藿定B:Y=19 818 696.4286X-17 005.0000,R2=0.9999,線性范圍0~0.28 mg·mL-1。
朝藿定C:Y=17 023 549.2611X+24 658.8571,R2=0.9991,線性范圍0~0.29 mg·mL-1。
寶藿苷Ⅰ :Y=29 157 312.4286X+20 219.6640,R2=0.9998,線性范圍0~0.28 mg·mL-1。
各對照品校準曲線見圖1,混合對照品及空白對照色譜圖見圖2。
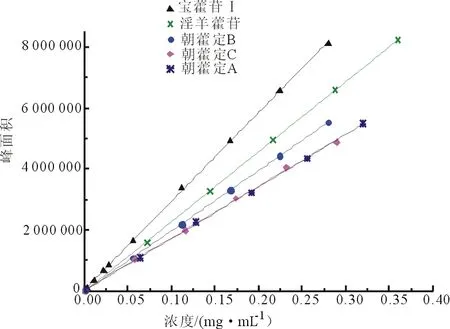
圖1 淫羊藿樣品含量測定用對照品校準曲線
2.2 含量測定結果
淫羊藿藥材不同部位HPLC分析色譜圖見圖2,淫羊藿藥材不同部位有效成分含量測定結果表3、圖3。
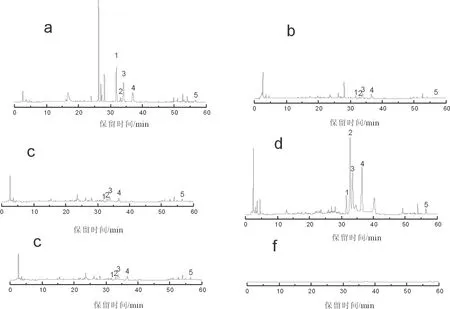
a.淫羊藿根; b.淫羊藿莖; c.淫羊藿葉柄; d.淫羊藿葉片; e.混合對照品; f.空白對照.1.朝藿定A; 2.朝藿定B; 3.朝藿定C; 4.淫羊藿苷; 5.寶藿苷Ⅰ
圖3 8批淫羊藿樣品不同部位中5種有效成分含量平均值比較
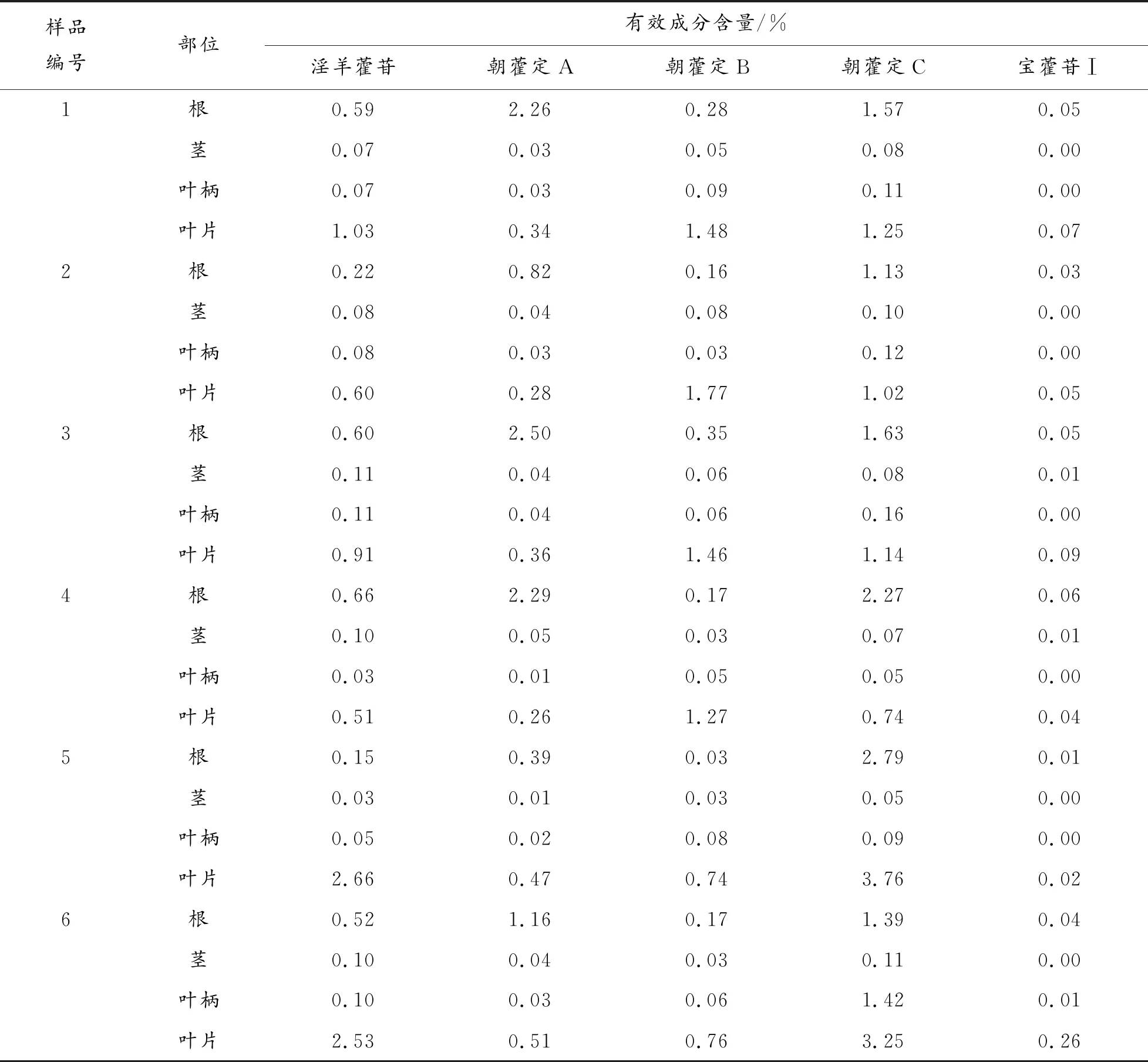
表3 淫羊藿藥材不同部位有效成分含量測定結果
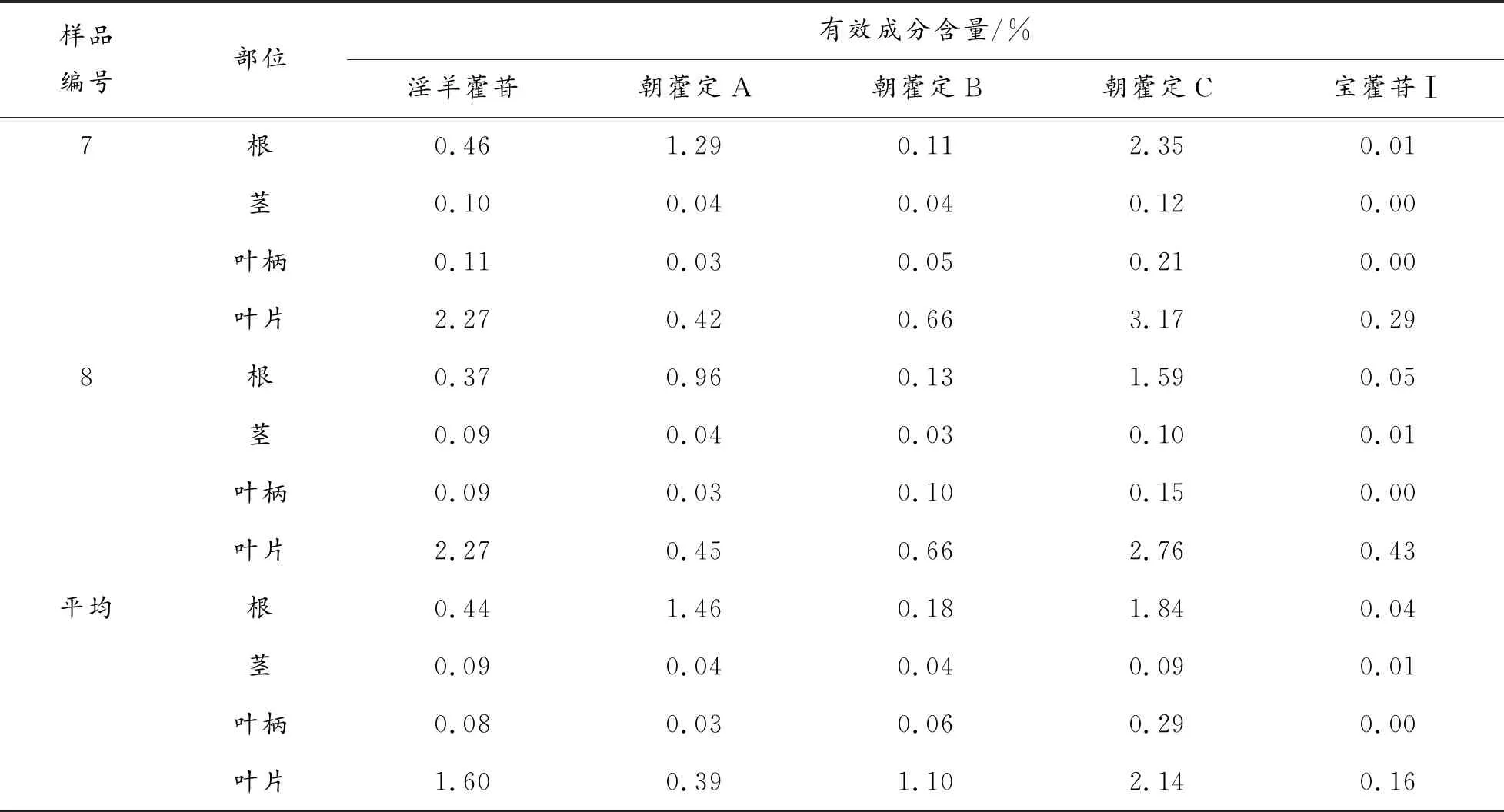
續表3 淫羊藿藥材不同部位有效成分含量測定結果
由圖2可知,空白對照色譜圖的基線平穩,對樣品分析無干擾。相較于淫羊藿根和淫羊藿葉片,淫羊藿莖、淫羊藿葉柄色譜圖中色譜峰的數目少、峰面積小,表明淫羊藿此2個部位中含有的化學物質較少。淫羊藿根、淫羊藿葉片色譜圖中色譜峰的數目更多、峰面積更大,表明淫羊藿根、葉片兩個部位中含有更豐富的化學物質,有效成分的含量更高。淫羊藿根樣品色譜圖在保留時間26~28 min間,出現了3個較為明顯,且在其它樣品色譜圖中未出現的色譜峰,表明淫羊藿根中的化學成分種類與其它部位不同,淫羊藿根與淫羊藿葉片的藥理作用有區別。
由表3、圖3可知,淫羊藿苷、朝藿定A、朝藿定B、朝藿定C及寶藿苷Ⅰ 5種成分在淫羊藿藥材不同部位中的含量差異較為明顯,其中淫羊藿葉片中有效成分含量最豐富,有4種成分的含量在各部位樣品中最高,淫羊藿苷、朝藿定A、朝藿定B、朝藿定C 4種成分含量之和高于藥典"淫羊藿、柔毛淫羊藿、箭葉淫羊藿均不得少于1.5%"的規定[7]。其次為淫羊藿根,朝藿定A在根中的含量最高;根中前述4種有效成分的含量之和,也能達到2020年版藥典的要求;淫羊藿莖、淫羊藿葉柄中5種有效成分的含量均較低,幾乎沒有藥用價值;寶藿苷Ⅰ的含量,在淫羊藿藥材各部位中的含量均較低。上述檢測結果表明,藥典關于淫羊藿藥用部位為"干燥葉",且生品藥材中不檢測寶藿苷Ⅰ的相關規定具有一定的合理性。
3 結論與討論
研究中發現以2020版藥典淫羊藿項目下規定的色譜條件,未能使朝藿定A、朝藿定B、朝藿定C以及淫羊藿苷等4種成分獲得良好分離。為解決上述問題,本文在新藥典規定方法的基礎之上,將樣品分析的色譜條件進行了調整。在調整后的色譜條件下,上述4種成分色譜峰間的分離度均可達1.5以上,各色譜峰的理論塔板數均在2.5×104以上。
多年來,淫羊藿藥材生產中一直視淫羊藿原植物的根、莖、葉柄等部位為雜質,造成了一定程度上的資源浪費,也增加了生產過程中能源及人力資源的消耗。為了驗證淫羊藿藥用部位劃分的合理性,本研究對淫羊藿不同部位中有效成分的含量進行了對比分析。由研究結果可以看出,淫羊藿莖、葉柄中幾乎不含有效成分,在生產中應作為雜質除去,以增加藥材品質。但淫羊藿根中化學物質含量豐富,特別是幾種藥典規定的指標性成分的含量之和可以達到標準要求,表明淫羊藿根具有一定的藥用開發價值。課題組在今后的工作中,將重點對淫羊藿根的藥理、藥效作用進行研究,探明其作為淫羊藿藥材使用的可行性,為拓寬淫羊藿藥用資源奠定基礎。
