氯氟吡啶酯水解產物的鑒定及其檢測方法的建立
2021-06-21 05:59:58屠王滿措馬有寧潘九月黃思琦陳銘學
農產品質量與安全 2021年3期
屠王滿措 馬有寧 潘九月 黃思琦 陳銘學
(中國水稻研究所農業農村部稻米及制品質量監督檢驗測試中心,農業農村部稻米產品質量安全風險評估實驗室,杭州310006)
水稻是我國第一大糧食作物,保證其產量與品質意義重大,而稻田雜草是影響水稻產量的主要原因之一[1~3]。除草劑具有效率高、效果好、成本低等優點,是現代農田雜草防除的主要手段[4]。然而農藥不管以何種方法施用,都不可避免農藥親體及其降解產物在環境中的遷移、轉化和積累[5]。農藥在環境中通過各種方式進行降解,其中水解是許多農藥降解的主要方式之一,同時也是評價農藥在環境中穩定性的一個重要指標[6]。由于很多農藥親體及其代謝產物在環境中的富集作用,對非靶標生物和整個生態系統存在很大的潛在危害[7]。因此,農藥在環境中的遷移、轉化及殘留檢測是農產品質量安全評估的主要內容之一[8]。
氯氟吡啶酯(4-氨基-3-氯-6-(4-氯-2-氟-3-甲氧基苯基)-5-氟吡啶-2-羧酸苯甲)(florpyrauxifen-benzyl)是全新芳香基吡啶甲酸酯類水稻除草劑,其主要通過與植物體內激素受體結合,干擾植物正常的生理生化功能,從而致目標植物死亡[9]。因用量少、復配性強等特點,近幾年氯氟吡啶酯被廣泛使用于稻田環境[10]。目前國內外對氯氟吡啶酯的研究報道大多集中在水稻雜草防治效果方面的研究[11~13],如倪青等[14]研究了氯氟吡啶酯對直播稻及移栽稻田雜草的防除效果,李建群等[15]研究了氯氟吡啶酯及其他農藥混配使用對稻田雜草的防效及水稻安全性方面的影響,結果表明,氯氟吡啶酯對水稻闊葉類雜草有很好的防治效果。然而對氯氟吡啶酯在稻田環境中的降解產物、殘留檢測及其在環境中的消解動態等方面還沒有相關研究報道,因此,研究氯氟吡啶酯在環境中的降解產物及消解動態對其在稻田環境的使用安全性評估十分必要。本實驗依據農業行業標準[16]進行,利用高效液相色譜-四極桿-飛行時間質譜(HPLC-QTOF/MS)技術鑒定氯氟吡啶酯在不同pH緩沖溶液中的水解產物,同時優化QuEChERS前處理方法,結合高效液相色譜串聯三重四極桿質譜技術建立了同時檢測稻田環境中氯氟吡啶酯及其代謝產物的方法,并且通過土培模擬試驗,動態監測氯氟吡啶酯在稻田環境中的代謝與遷移,為氯氟吡啶酯在稻田生態系統中的安全性評估提供參考依據。
一、 材料與方法
(一)試驗材料
1.材料與試劑。氯氟吡啶酯(florpyrauxifen-benzyl,純度98.6%)和氯氟吡啶酸(florpyrauxifen,純度98.5%)標準品,德國Dr.Ehrenstorfer LGC公司;乙腈(色譜純,德國Merck公司);甲酸(色譜純,美國Tedia公司);甲醇(色譜純,德國Merck公司);無水硫酸(MgSO4,分析純)和氫氧化鈉(NaOH, 分析純,Sigma-Aldrich公司);氯化鈉(NaCl,分析純,上海試四赫維化工有限公司);硼酸(H3BO3,分析純,太倉美達試劑有限公司);氯化鉀(KCl,分析純,寧波市化學試劑廠);檸檬酸鉀(C6H5K3O7,分析純);磷酸二氫鉀(KH2PO4,分析純,上海沃凱生物技術有限公司);乙二胺-N-丙基硅烷(PSA,美國瓦里安公司),十八烷基鍵合硅膠吸附劑(C18,美國瓦里安公司)和石墨化炭黑(GCB,德國Thermo Fisher Scientific公司),粒徑40μm。
2.儀器與設備。HPLC-Q-TOF/MS,美國Agilent公司;Surveyor液相色譜(美國Thermo Fisher Scientific公司);Poroshell 120 EC-C18色譜柱(150 mm×2.1mm,2.7μm,美國Waters公司);TSQ Quantum Access Max三重四極桿質譜儀(德國Thermo Fisher Scientific公司);分析天平(瑞士Mettler toledo公司);Milli-Q Advangtage超純水儀(美國Millipore公司);PHS-3C精密pH計(上海儀電科學儀器股份有限公司);恒溫恒濕箱(上海博迅實業有限公司);T 25基本型高速勻漿機(德國IKA公司);Primo R臺式離心機(德國Thermo Fisher Scientific公司)。
(二)溶液的配制分別稱取10 mg(精確至0.1 mg)氯氟吡啶酯和氯氟吡啶酸標準品,用丙酮溶液溶解并定容至10 mL,配成濃度為1 000 mg/L的標準儲備液。分別吸取標準儲備液各1 mL,于10 mL容量瓶中用乙腈稀釋至刻度,配制氯氟吡啶酯和氯氟吡啶酸濃度為100 mg/L混合中間液。準確吸取中間液1 mL,于10 mL容量瓶中用乙腈稀釋至刻度,配成濃度為10 mg/L的標準工作液,均避光-20℃保存。將標準工作液用乙腈稀釋成濃度分別為500、200、100、50和5μg/L的上機液,現用現配。
(三)實驗內容
1.HPLC-Q-TOF/MS鑒定氯氟吡啶酯水解產物。
(1)緩沖溶液的配制。根據農業行業標準NY/T 1860.9-2016《農藥理化性質測定試驗導則》配制pH 9、pH 7和pH 4的緩沖溶液。配制的所有緩沖溶液和所用的玻璃容器均在高溫高壓滅菌鍋中滅菌處理,用滅菌后的緩沖溶液將1 000 mg/L的氯氟吡啶酯標準品稀釋配制成200 mL 1.0 mg/L的溶液,配制好的處理液分別于3個100 mL三角瓶中密封,放置于50℃恒溫恒濕培養箱中進行黑暗保存。另取200 mL滅菌好的緩沖溶液分別置于3個100 mL三角瓶中作為空白對照,同上條件保存,于培養開始后的第7 d取樣測定。
(2)樣品提取。準確量取20.0 mL水樣于150.0 mL離心管中,向離心管中加入25.0 mL乙腈,高速勻漿(10 000 r/min)1 min,加入10.0 g無水MgSO4和1.0 g NaCl再次高速勻漿(10 000 r/min)1 min,以3 500 r/min離心3 min,取上清液1.0 mL過0.22μm有機相濾膜待測。
(3)儀器方法。色譜條件:色譜柱為XSelect HSS T3色譜柱,柱溫45℃。含0.1%甲酸水溶液(V/V)(A)和乙腈(B)作為流動相,梯度洗脫0~5 min,5%~35%B;5~10 min,35%~98%B;10~17 min,98%B;17~20 min,98%~5%B;20~25 min,5%B;流速為0.3 mL/min,進樣量為5μL。
質譜采集條件:離子源為電噴霧ESI源,采用正離子全掃描模式,質量掃描范圍m/z50~1 000,一級質譜掃描速率為5 spectra/s。質譜參數設置為干燥氣溫度350℃,干燥氣流速8 L/min;鞘氣溫度280℃,鞘氣流速11 L/min;碎裂電壓130 V;霧化氣壓力40 psi;射頻電壓750 V;錐孔電壓40 V;噴嘴電壓500 V;毛細管電壓4 000 V。在采集數據時,通過注入參比離子對儀器進行質量精度實時校正。校正離子為嘌呤(m/z121.050873)和HP-0921(m/z922.009798)。二級掃描碰撞能量20 eV,掃描速率為3 spectra/s,其他同一級質譜條件。
(4)數據采集與分析。數據采集和系統控制由Agilent HPLC-Q-TOF/MS MassHunter采集軟件(B.06.00)和定性分析軟件(B.07.00)完成。通過Agilent MassHunter Profinder軟件(10.0) 對色譜峰進行峰匹配、峰提取、峰對齊、峰識別、基線矯正以及歸一化進行處理,然后通過Agilent MassProfiler Professional(MPP)軟件進行頻數過濾、Ttest(P<0.05) 顯著分析和倍數(FC>2)比較進行差異化分析。為了確定代謝物的結構,針對一級質譜篩選到的6個差異化合物進行二級質譜數據的采集,通過Agilent LC-Q-TOF/MS MassHunter(B.07.00)軟件進行化合物提取并導出CEF文件格式,然后導入MassHunter Molecular Structure Correlator(MSC)軟件進行定性分析。
2.氯氟吡啶酯及其代謝物檢測方法的建立。
(1)樣品的提取。稻田水:量取20.0 mL稻田水于200.0 mL離心管,加入25.0 mL含0.1%的甲酸乙腈(V/V),經高速勻漿1 min后,加入1.0 g NaCl和10.0 g MgSO4后再次高速勻漿,以3 500 r/min離心3 min,取上清液1 mL過0.22μm有機濾膜待HPLC-MS/MS檢測。
水稻苗(根部、地上部):分別稱取5.0 g(精確至0.01g)樣品于250.0 mL離心管,加入20.0 mL超純水充分浸泡30 min,加入25.0 mL含0.1%的甲酸乙腈(V/V),經高速勻漿后2 min,加入1.0 g NaCl和10.0 g MgSO4后再次高速勻漿,以3 500 r/min離心3min。取上清液5 mL待凈化。
土壤(60℃烘3 h):分別稱取5.0 g(精確至0.01g)樣品于200.0 mL離心管,加入20.0 mL超純水充分浸泡30 min,加入25.0 mL含0.1%的甲酸乙腈(V/V),經搖床振蕩30 min后超聲提取30 min,加入1.0 g NaCl和10.0 g MgSO4后再次超聲提取15 min,以3 500 r/min離心3 min。取上清液5 mL待凈化。
(2)樣品的凈化。取上述上清液5 mL于100 mg C18、50 mg GCB和1.2 g無水MgSO4的混合物,渦旋混合器震蕩1 min,充分混勻后,以3 800 r/min離心5 min,取上清液1 mL過0.22μm有機濾膜待HPLC-MS/MS檢測。
(3)儀器條件。色譜條件:色譜柱為Poroshell 120 EC-C18(150×2.1 mm,2.7μm) 柱, 流動相為0.1%甲酸水-乙腈溶液(V/V)(0.1%甲酸水為A相,乙腈為B相,梯度洗脫設置為0~5 min,40%~80%B;5~7 min,80%~95%B;7~10 min,95%B;10~10.1 min,95%~40%B;10.1~15 min,40%B; 流速200μL/min, 進樣量為2μL。化合物保留時間見表1。
質譜條件:質譜所用電噴霧源為ESI,采用正離子掃描模式,動態多反應監測,離子源溫度為300℃,毛細管溫度為350℃,鞘氣(N2)壓力為50 Arb,輔助氣(N2)壓力為20 Arb,噴霧電壓為3 500 V(ESI+)。其他譜參數見表1。

表1 化合物的動態多反應監測質譜采集離子信息
3.氯氟吡啶酯及代謝物氯氟吡啶酸在稻田環境中的消解動態。
(1)土壤的預處理。選取中國水稻研究所富陽基地未曾施用氯氟吡啶酯農藥的土壤,風干過1 mm(16目)的篩子,每份約30 kg土壤裝于塑盆(60 cm×40 cm×20 cm)中,充分淹水處理48 h待移栽水稻幼苗。
(2)水稻幼苗的培養。水稻品種選用中浙優1號,將種子用2%的NaClO充分浸泡48 h后用蒸餾水反復沖洗,在35℃、空氣濕度85%的恒溫箱催芽24 h,催好芽的種子轉移至秧盤基質進行育秧25 d,將長勢一致的秧苗移栽至上述處理好的(60 cm×40 cm×20 cm)塑料盆中,每盆移栽120株,移栽后的秧苗進行7 d的恢復培養。
秧苗在塑料盆恢復處理7 d后進行施藥處理,參照農藥登記信息網推薦的施藥劑量和施用方法,以氯氟吡啶酯推薦施藥劑量的2倍(0.240 g(a.i.)/667 m2)進行試驗,施藥后保水5~7 d,水層保持5 cm,處理組和對照組分別設置3個重復。土培試驗于水稻研究所富陽基地的大棚中進行,分別于處理2 h和2、3、5、7、14、21、28 d進行取樣。
(3)樣品的采集及預處理。隨機取水稻幼苗(地上部、根部)5株經剪碎,淺層田水50 mL充分混勻,淺層土壤(5 cm)3個點混勻,經60℃烘箱烘干。所有樣品保存在-20℃的冰柜。
二、 結果與分析
(一)HPLC-Q-TOF/MS鑒定氯氟吡啶酯水解產物
1.分析方法穩定性驗證。在每次樣品采集的過程中進行質控樣品的采集,以避免儀器誤差引起實驗結果的干擾,確保分析系統的穩定性[17]。質控樣品的色譜如圖1所示,可以看出,質控樣品采集的色譜峰重現性良好,可以確保分析過程中儀器系統具有較好穩定性和實驗數據結果的可靠性。

圖1 質控樣品的色譜疊加圖(n=4)
2.鑒定結果分析。實驗經處理7d后,利用LC-Q-TOF/MS進行數據采集,借助MPP軟件對數據進行頻數過濾、Ttest(P<0.05)顯著分析及倍數比較(FC>2)差異化分析,結果在3組不同的pH處理組中篩選到6個差異化合物,其質荷比m/z值和保留時間如圖2所示,其中質荷比m/z值439.042和367.042的2個化合物出峰時間較為接近,均在11.86±0.02 min出峰,而后4個化合物質荷 比m/z值 348.995、 330.848、 302.9896 和268.0243的出峰時間較接近,均在9.53±0.004 min出峰。

圖2 處理組中6個差異化合物的質荷比m/z、出峰時間及提取離子色譜圖
為了明確中間代謝物的結構,在碰撞能量20 eV的條件下對篩選到的6個差異化合物進行了二級譜圖掃描。經MSC軟件搜庫檢索,最終鑒定到2個化合物,具體信息見表2,其中保留時間為11.864 min,質荷比m/z439.042為氯氟吡啶酯,而保留時間為9.544 min,質荷比m/z348.995為氯氟吡啶酸,其質量數偏差分別為1.46和0.8 mDa。為了保證結果的可靠,將樣品中鑒定到的氯氟吡啶酯及氯氟吡啶酸與其標準品的出峰時間及二級譜圖(見圖3~圖6)進行確證比對,結果出峰時間完全一致。其他4個差異化合物中質荷比m/z367.042與氯氟吡啶酯的出峰時間相差0.02 min,與標準品碎片離子質荷比m/z值367.042的相對離子豐度和質荷比十分接近,其質量數偏差0.32 mDa,匹配度達到99.6分。而質荷比m/z330.988、302.988和268.024幾個離子與氯氟吡啶酸的出峰時間相差±0.004 min,與標準品碎片離子的相對離子豐度和質荷比十分匹配,其質量數偏 差分別為5.6、2.6和2.8 mDa,匹配度分別達到99.9、95.7及98.2分。因此推測其他4個差異化合物是氯氟吡啶酯及氯氟吡啶酸由于源內裂解所產生的碎片離子。

表2 氯氟吡啶酯和代謝物氯氟吡啶酸的基本信息

圖3 氯氟吡啶酯在樣品(A、C)及標準品(B、D)中的二級色譜圖、質譜圖
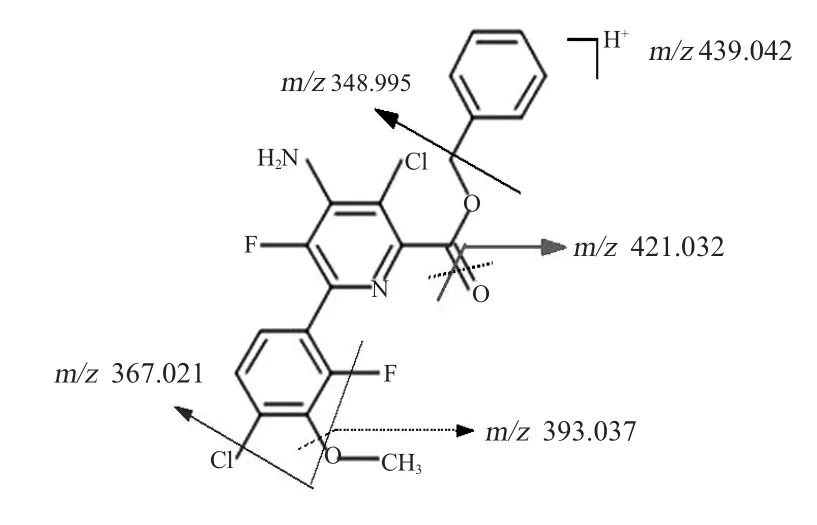
圖4 氯氟吡啶酯碎片斷裂的可能途徑

圖5 氯氟吡啶酸在樣品(A、B)及標準品(C、D)中的二級色譜圖、質譜圖

圖6 氯氟吡啶酸碎片斷裂的可能途徑
(二)氯氟吡啶酯及代謝物氯氟吡啶酸檢測方法的建立
1.提取溶劑的優化。氯氟吡啶酯及代謝物氯氟吡啶酸均為酸性化合物,提取溶劑中加入適量的酸,能夠抑制化合物的電離,使酸性化合物保持相對穩定,有利于提高化合物的回收率。因此分別向提取溶劑乙腈中加入體積為0%、0.1%、0.5%、1%和2%的甲酸以確定最佳提取溶劑。
本試驗通過在水稻苗根部、地上部,土壤和稻田水空白基質中的加標水平均為50μg/kg(水50 μg/L),經不同溶劑提取后,氯氟吡啶酯及氯氟吡啶酸的提取回收率如圖7所示,由圖可知,乙腈中甲酸的含量對氯氟吡啶酯的提取回收率影響不大,回收率均在85%以上。隨著甲酸含量的增加,氯氟吡啶酸的提取效率顯著增加,當甲酸含量達到0.1%時,回收率可達到97%,因此,選擇含有0.1%甲酸乙腈(V/V)溶液作為提取溶劑。

圖7 提取溶劑中不同甲酸的含量對氯氟吡啶酯(A)和氯氟吡啶酸(B)提取回收率的影響
2.吸附劑種類及用量的優化。GCB、C18及PSA對基質中甾醇類、花青素、糖類以及部分極性色素具很強的凈化能力。然而,吸附劑的種類和添加量都會影響目標化合物的回收率[18~19]。因此考察了GCB、C18及PSA對氯氟吡啶酯及代謝物氯氟吡啶酸的回收率。本實驗中2個化合物的加標水平為50μg/kg(水50μg/L),回收率結果見圖8。GCB添加劑量為0~50 mg時2個化合物的回收率達到83.6%~103.4%,但是隨著添加量的增加,對2個化合物都有明顯的吸附作用,吸附率達到30%以上。當C18的添加量為0~200 mg時對2個目標化合物都沒有明顯的吸附作用,2個化合物的回收率為91.3%~105.6%。當C18的添加量為0~100 mg時,2個目標化合物的基質效應從22%減弱到15%。當PSA添加量為0~200 mg時,氯氟吡啶酯的回收率為97.2%~100.4%,沒有吸附作用,而對代謝物氯氟吡啶酸有很強的吸附作用,吸附率達到90%。所以選用50 mg GCB、100 mg C18和1.2 g無水MgSO4時凈化劑效果最佳,2個目標化合物回收率達到83.6%~105.6%,基質效應<15%,滿足農藥檢測的要求。
3.方法學評價。
(1)定量限、檢出限、線性范圍及基質效應。經儀器優化后的條件下測定用初始流動相配制一系列不同濃度的2種化合物的混合標準溶液,以分析物的濃度為橫坐標(x,μg/L),定量離子對的響應值為縱坐標(y峰面積),利用外標法進行定量,獲得線性方程和相關系數見表3。結果表明,2種化合物在0.5~500.0μg/L線性范圍內,化合物的質量濃度與相應的響應值呈較好線性關系,線性相關系數在0.998以上。空白樣品中添加2種化合物的標準混合溶液,以S/N=10確定方法的定量限(LOQ),在稻田環境4種基質中氯氟吡啶酯及代謝物氯氟吡啶酸的定量限分別是2.5μg/kg和5.0 μg/kg,兩種目標化合在稻田環境4種基質中的基質效應為-0.4%~15.0%。

圖8 3種凈化劑的添加量對氯氟吡啶酯(A)及氯氟吡啶酸(B)回收率的影響

表3 化合物的檢出限、定量限、線性方程及其相關系數和基質效應(n=3)
(2)方法的回收率與精密度。分別稱取水稻苗地上部、根部、土壤以及稻田水空白樣本進行加標回收試驗,每個處理6個重復,對兩個化合物在不同添標水平方法的準確度和精密度進行考察。測定結果見表4,可以看出,兩種化合物在不同的基質中回收率為89.7%~110.4%,相對標準偏差(RSD)≤10%,回收率和精密度良好,滿足SANTE/11945/2015[20]的要求。
(三)氯氟吡啶酯及代謝物氯氟吡啶酸在稻田環境中的消解動態通過土培試驗對氯氟吡啶酯和代謝物氯氟吡啶酸在稻田環境中的含量進行動態監測,結果見表5~表6,結果表明,氯氟吡啶酯和代謝物氯氟吡啶酸在稻田環境中降解動態滿足一級動力學方程。氯氟吡啶酯在稻田水、地上、根部及土壤基質中半衰期分別為0.3、1.1、1.1和1.2 d,其在稻田水中降解速率最快,施藥5 d后降解達到90%,而在植株體和土壤介質中施藥21 d后降解率達到90%。代謝物氯氟吡啶酸施藥2 h后就出現在稻田環境中,其在稻田水、水稻苗地上、水稻苗根部及土壤基質中的半衰期分別為3.4、1.6、2.4和2.7 d,其在水稻苗地上部降解最快,施藥后21 d降解率達到80%。通過參照 《化學農藥環境安全評價試驗準則》(2008)中農藥的環境行為特性等級劃分,氯氟吡啶酯和代謝物氯氟吡啶酸在稻田環境各介質中的半衰期T1/2<1(月),屬于易降解農藥。

表5 氯氟吡啶酯在稻田環境中的消解動態

表6 氯氟吡啶酸在稻田環境中的消解動態
三、結論
本文利用HPLC-Q-TOF/MS高分辨質譜鑒定、標準品確證等手段研究了氯氟吡啶酯的水解產物,結果發現,氯氟吡啶酯在水中主要降解為氯氟吡啶酸。通過優化QuEChERS方法提取溶劑的pH值及3種凈化劑(GCB、PSA、C18)的添加劑量,結合高效液相色譜串聯三重四極桿質譜技術建立了同時測定稻田中氯氟吡啶酯及其代謝物的方法。結果表明,當方法以0.1%甲酸乙腈(V/V)作為提取溶劑,50 mg GCB、100 mg C18和1.2 g無水MgSO4作為凈化劑,2種化合物在4種不同基質中的回收率為89.7%~110.4%,基質效應為-0.4%~15.0%,相對標準偏差≤10%。說明該方法具有較高的精密度、靈敏度高、重現性好,可進行準確的定性定量分析。通過土培試驗結果表明,氯氟吡啶酯在稻田環境中富集能力為地上部>根部>稻田水>土壤,水稻苗地上部、水稻苗根部和稻田水在施藥2 h后達到最高值,隨后迅速降解;代謝物氯氟吡啶酸在施藥2 h后即出現在稻田環境里,主要分布在植株和稻田水中。氯氟吡啶酯與代謝物氯氟吡啶酸在稻田環境中半衰期<1(月),施藥后28 d在稻田壞境中降解率達到90%以上,屬于易降解農藥。
