食管癌化療抗性的分子機(jī)制
2021-07-03 09:24:40許麗艷衡晶華李恩民
食管疾病 2021年2期
許麗艷,衡晶華,李恩民
目前,治療食管癌的化療藥物主要是鉑類(lèi)和氟尿嘧啶類(lèi)。在患者一般狀況較好的情況下,也可使用紫杉醇或阿霉素等[1]。不同的化療藥物有不同的作用機(jī)理。順鉑(Cisplatin,DDP)通過(guò)與DNA分子中的嘌呤堿基上的N7反應(yīng),形成加合物,破壞細(xì)胞DNA的結(jié)構(gòu),干擾DNA修復(fù),誘導(dǎo)癌細(xì)胞凋亡[2]。5-氟尿嘧啶(5-fluorouracil,5-FU)的細(xì)胞毒性機(jī)制歸因于5-FU誤摻入RNA和DNA分子中,以及對(duì)胸苷酸合成酶(thymidylate synthase,TS)的抑制。5-FU在細(xì)胞內(nèi)轉(zhuǎn)化為幾種活性代謝物:氟脫氧尿苷一磷酸酯(fluorodeoxyuridine monophosphate,F(xiàn)dUMP)、氟脫氧尿苷三磷酸(fluorodeoxyuridine triphosphate,F(xiàn)dUTP)和氟尿嘧啶三磷酸(fluorouridine triphosphate,F(xiàn)UTP),它們均可破壞RNA和DNA的合成,以及抑制TS。5-FU分解代謝的限速酶是二氫嘧啶脫氫酶(dihydropyrimidine dehydrogenase,DPD),可將5-FU轉(zhuǎn)化為二氫氟尿嘧啶(dihydrofluorouracil,DHFU)[3]。紫杉醇通過(guò)促進(jìn)微管蛋白的聚合,阻止其解聚,發(fā)揮穩(wěn)定微管的作用,抑制細(xì)胞微管的動(dòng)態(tài)重組,將細(xì)胞周期阻滯于G2/M期,阻礙細(xì)胞分裂,誘發(fā)細(xì)胞死亡[4]。然而,化療藥治療食管癌會(huì)產(chǎn)生各種耐藥,導(dǎo)致對(duì)藥物的應(yīng)答率降低。本文從靶前耐藥、靶上耐藥和靶后耐藥等不同角度,闡述細(xì)胞對(duì)DDP、5-FU以及紫杉醇的抗性機(jī)制。
1 靶前耐藥
ABC(ATP binding cassette)轉(zhuǎn)運(yùn)蛋白家族,主要包括ABCB1基因編碼的MDR1(multidrug resistance 1),又稱(chēng)P-糖蛋白或P-gp(P-glycoprotein),以及MRP1(multidrug resistance associated protein 1)和ABCG2(ATP-binding cassette subfamily G member 2)等[5]。ABC家族主要通過(guò)增強(qiáng)藥物外排,降低細(xì)胞核內(nèi)DDP濃度,導(dǎo)致耐藥。SRPX2基因和FZD-7基因所編碼蛋白均還可通過(guò)Wnt/β-catenin信號(hào)通路促進(jìn)MDR1表達(dá),促進(jìn)食管鱗狀細(xì)胞癌(ESCC)細(xì)胞的增殖和侵襲轉(zhuǎn)移[6-7]。換言之,通過(guò)抑制MDR1可以逆轉(zhuǎn)DDP抗性。例如YB-1(Y-box-binding protein 1)可抑制MDR1的轉(zhuǎn)錄,增強(qiáng)DDP誘導(dǎo)的EC109和TE-1細(xì)胞的細(xì)胞毒作用[8];恩替司他通過(guò)抑制Src/Mcl-1/MDR1信號(hào)軸,顯著抑制DDP耐藥小鼠的腫瘤生長(zhǎng)[9]。從中草藥“漢防己”中分離出來(lái)的漢防己堿(tetrandrine,TET)可抑制MDR1的表達(dá)和活性,干預(yù)耐藥的發(fā)生[10]。另外,在TE-5R內(nèi)發(fā)現(xiàn)DPD過(guò)表達(dá)導(dǎo)致5-FU的快速降解,DPD抑制劑吉美拉西可顯著增加TE-5R細(xì)胞內(nèi)5-FU濃度,減弱5-FU耐藥性[11]。THUMPD2基因下調(diào)表達(dá)引起藥物外排增多,進(jìn)而導(dǎo)致DDP和5-FU抵抗[12]。相關(guān)情況見(jiàn)圖1,ABC蛋白家族促進(jìn)藥物外排,降低核內(nèi)藥物濃度。
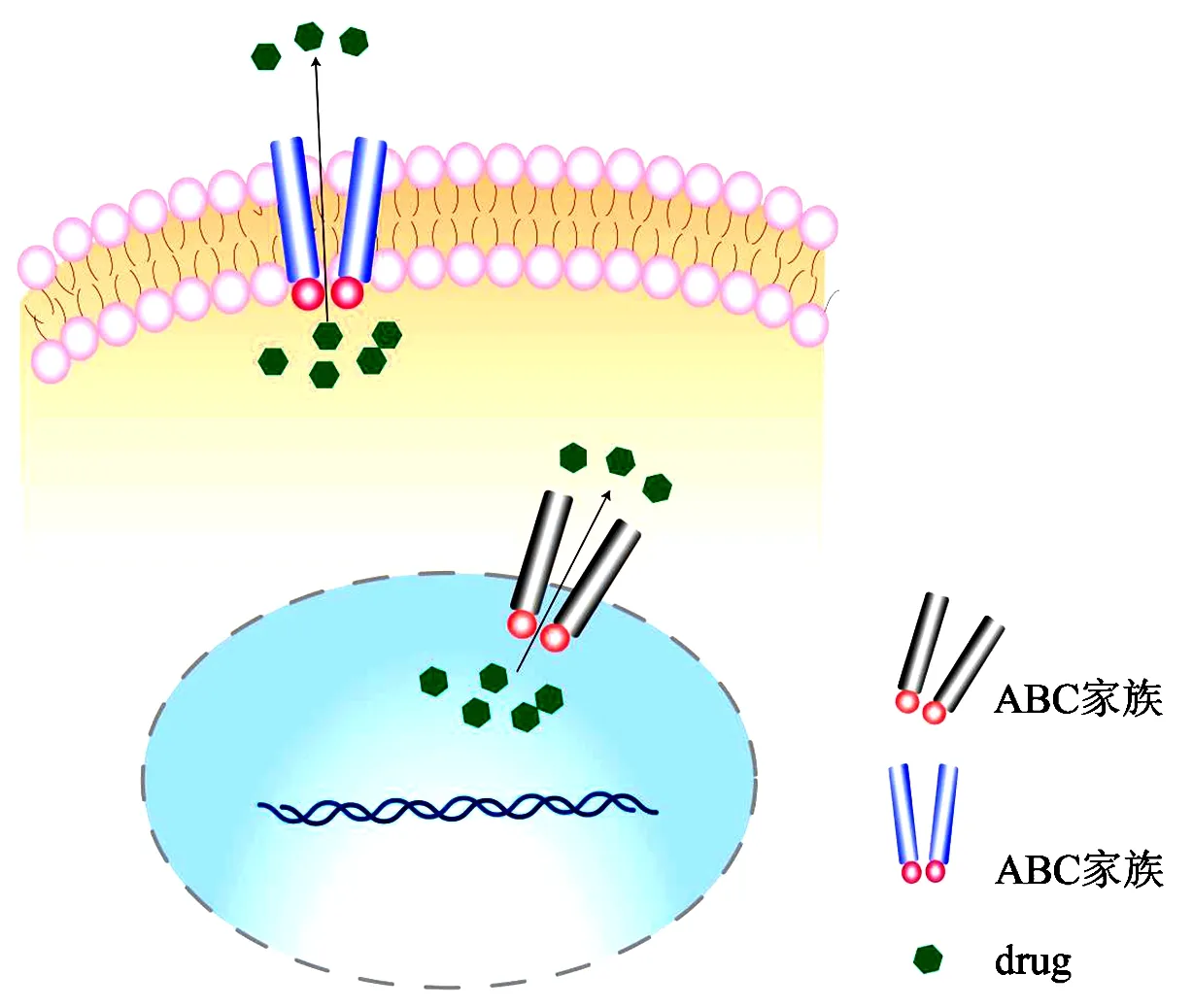
圖1 靶前耐藥模式圖
2 靶上耐藥
2.1 DNA損傷修復(fù)介導(dǎo)DDP抗性
如果DNA損傷發(fā)生在有絲分裂前,并且無(wú)法修復(fù),就會(huì)誘發(fā)細(xì)胞凋亡。因此,DNA損傷修復(fù)(DNA damage repair,DDR)是腫瘤細(xì)胞逃避因化療藥物毒性引起細(xì)胞死亡的重要的耐藥機(jī)制。
2.1.1 DNA損傷修復(fù)途徑損傷修復(fù)方式包括堿基切除修復(fù)(base excision repair,BER)、錯(cuò)配修復(fù)(mismatch repair,MMR)、同源重組(Homologous Recombination,HR)、非同源末端連接(nonhomologous end joining,NHEJ)等。①BER:鉑類(lèi)化合物誘導(dǎo)細(xì)胞產(chǎn)生的活性氧自由基(reactive oxygen species,ROS),可導(dǎo)致脂質(zhì)、蛋白質(zhì)、胞核DNA和線(xiàn)粒體DNA(mitochondrial DNA,mtDNA)損傷。其中,最穩(wěn)定且有害的ROS修飾產(chǎn)物之一是8-羥基鳥(niǎo)嘌呤(8-Hydroxyguanine,8-oxoG),該損傷修復(fù)通過(guò)BER進(jìn)行。其中,hOGG1-1a(a-hOGG1)主要修復(fù)核DNA損傷,hOGG1-2a(b-hOGG1)主要修復(fù)mtDNA損傷,介導(dǎo)DDP抗性[13]。②MMR:XPC是一種重要的DNA損傷識(shí)別蛋白,它不僅參與核苷酸切除修復(fù)(nucleotide excision repair,NER),還參與MMR和雙鏈斷裂修復(fù)途徑[14]。③HR修復(fù):增強(qiáng)細(xì)胞損傷修復(fù)能力的BRCA1作為HR系統(tǒng)的關(guān)鍵功能蛋白,可以通過(guò)與BARD1形成異源二聚體參與DDR。整合素α-5通過(guò)粘著斑激酶FAK激活PI3K/AKT信號(hào)通路,作用于BARD1,激活DDR,抗細(xì)胞凋亡,介導(dǎo)DDP耐藥[15]。④NHEJ修復(fù):DNA依賴(lài)的蛋白激酶全酶(Ku70、Ku80和DNA-PKcs)是NHEJ的關(guān)鍵啟動(dòng)酶。HOXB7使Ku70、Ku80和DNA-PKcs表達(dá)上調(diào),將細(xì)胞周期阻滯于S期,產(chǎn)生DDP抗性[16]。
2.1.2 ATM和ATRATM(ataxia-telangiectasia mutated)和ATR(ataxia telangiectasia and Rad3-related)的激活對(duì)于DDR和維持基因組穩(wěn)定性至關(guān)重要,抑制ATM或ATR被認(rèn)為是一種很有前景的化療增敏策略。E2F1激活miR-26b的轉(zhuǎn)錄,其與ATM和RB基因的3UTR相互作用,下調(diào)ATM和RB的表達(dá),增強(qiáng)DDP對(duì)食管癌化療的敏感作用[17]。STAT3對(duì)于有效修復(fù)受損的DNA也是必不可少的。ATR的阻斷劑VE-822與DDP的聯(lián)合作用顯著抑制了食管癌KYSE450和KYSE70細(xì)胞中p-STAT3的表達(dá),特別是對(duì)ATM缺陷型細(xì)胞的增敏作用更加明顯,并且引起了包括Hippo通路、MAPK通路、JAK-STAT通路和PI3K-AKT通路的廣泛變化[18]。
2.1.3 其他定量蛋白質(zhì)組學(xué)結(jié)合生物信息學(xué)和蛋白功能分析發(fā)現(xiàn),RIP3通過(guò)上調(diào)HSP90-CDC37復(fù)合物和ERK、JNK和AKT激酶等下游信號(hào),以一種不依賴(lài)激酶的方式調(diào)節(jié)DNA修復(fù),導(dǎo)致DDP靶向耐藥[19]。
2.2 核苷酸代謝與5-FU抗性
核苷酸代謝中的關(guān)鍵酶如DPD上調(diào)表達(dá)和TS活性增加,參與了癌細(xì)胞對(duì)5-FU耐藥。ID-1與E2F1競(jìng)爭(zhēng)結(jié)合Cdc20,激活I(lǐng)GF2基因表達(dá),阻斷IGF1R(Insulin-like growth factor type 1 receptor),間接上調(diào)TS,介導(dǎo)5-FU耐藥[20]。miR-338-5p與ID-1編碼基因的mRNA的3’-UTR直接相互作用,抑制ID-1表達(dá),介導(dǎo)ESCC對(duì)5-FU增敏[21]。二甲雙胍誘導(dǎo)核苷酸代謝改變,如其中TS和胸苷激酶TK1(thymidine kinase 1)的表達(dá)增加可能導(dǎo)致細(xì)胞內(nèi)dTTP池的增加和5-FU合成代謝物的“稀釋”,介導(dǎo)5-FU耐藥[22]。胰島素通過(guò)增加5-FU攝取,增加胞內(nèi)游離TS、降低TS三元復(fù)合物,以及上調(diào)活性的Caspase-3的表達(dá),促進(jìn)對(duì)5-FU增敏[23]。細(xì)胞周期阻滯使胸腺嘧啶耗竭,介導(dǎo)5-FU增敏。REV3L表達(dá)下調(diào)通過(guò)抑制CyclinD1和Survivin的表達(dá),誘導(dǎo)G1期阻滯和凋亡,增加細(xì)胞對(duì)5-FU的敏感性[24]。過(guò)表達(dá)miR-145,靶向REV3L,顯著提高5-FU抑制抗凋亡經(jīng)典分子Bcl-2家族蛋白表達(dá),提高細(xì)胞凋亡率[25]。
2.3 抑制G2/M期阻滯介導(dǎo)紫杉醇耐藥
在多個(gè)紫杉醇耐藥的細(xì)胞系中,發(fā)現(xiàn)細(xì)胞周期分布改變:G0/G1和S期細(xì)胞增多,G2/M期細(xì)胞減少[26]。5-AZ可通過(guò)誘導(dǎo)細(xì)胞周期相位重新分布,使耐藥ESCC細(xì)胞對(duì)紫杉醇化療重新增敏[27-28]。STMN1(Stathmin)通過(guò)促進(jìn)微管解聚和/或防止微管蛋白異二聚體聚合,調(diào)節(jié)微管動(dòng)力學(xué);沉默STMN1基因表達(dá)可以通過(guò)G2/M期阻滯增加ESCC對(duì)紫杉醇的敏感性[29-30]。有關(guān)食管癌化療紫杉醇和5-FU的靶上耐藥機(jī)制見(jiàn)圖2,其中DNA損傷修復(fù)介導(dǎo)DDP抗性;TS、TK1增加胞內(nèi)dTTP池,降低5-FU合成代謝物濃度,介導(dǎo)5-FU抗性;解除G2檢查點(diǎn)介導(dǎo)紫杉醇抗性。
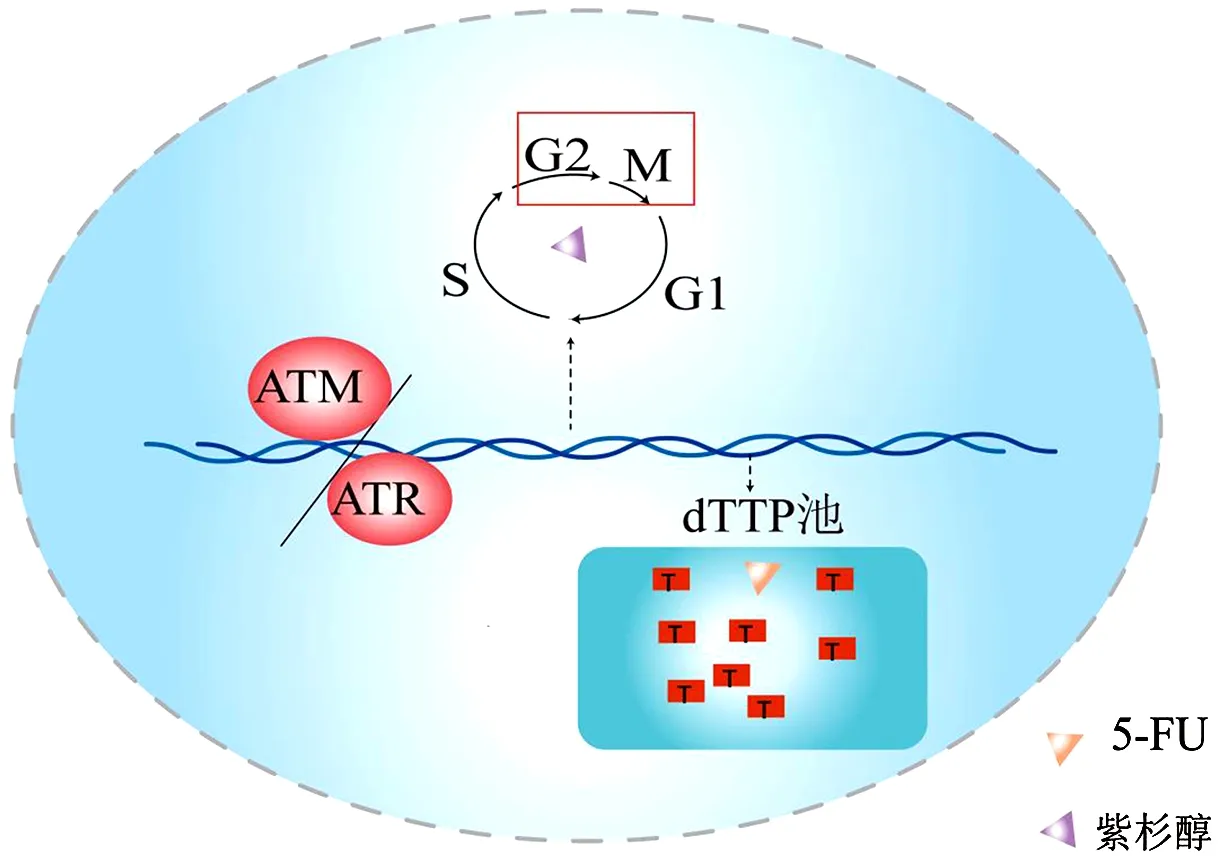
圖2 靶上耐藥模式圖
3 靶后耐藥
腫瘤細(xì)胞可以通過(guò)細(xì)胞程序性死亡系統(tǒng)的改變對(duì)化療藥物產(chǎn)生抵抗,涉及腫瘤細(xì)胞本身及腫瘤微環(huán)境(tumor microenvironment,TME)。有關(guān)食管癌靶后耐藥相關(guān)機(jī)制見(jiàn)圖3,促進(jìn)增殖、抑制凋亡、侵襲轉(zhuǎn)移、腫瘤細(xì)胞干性、Warburg效應(yīng)、CAFs分泌因子均介導(dǎo)不同化療藥物耐藥。
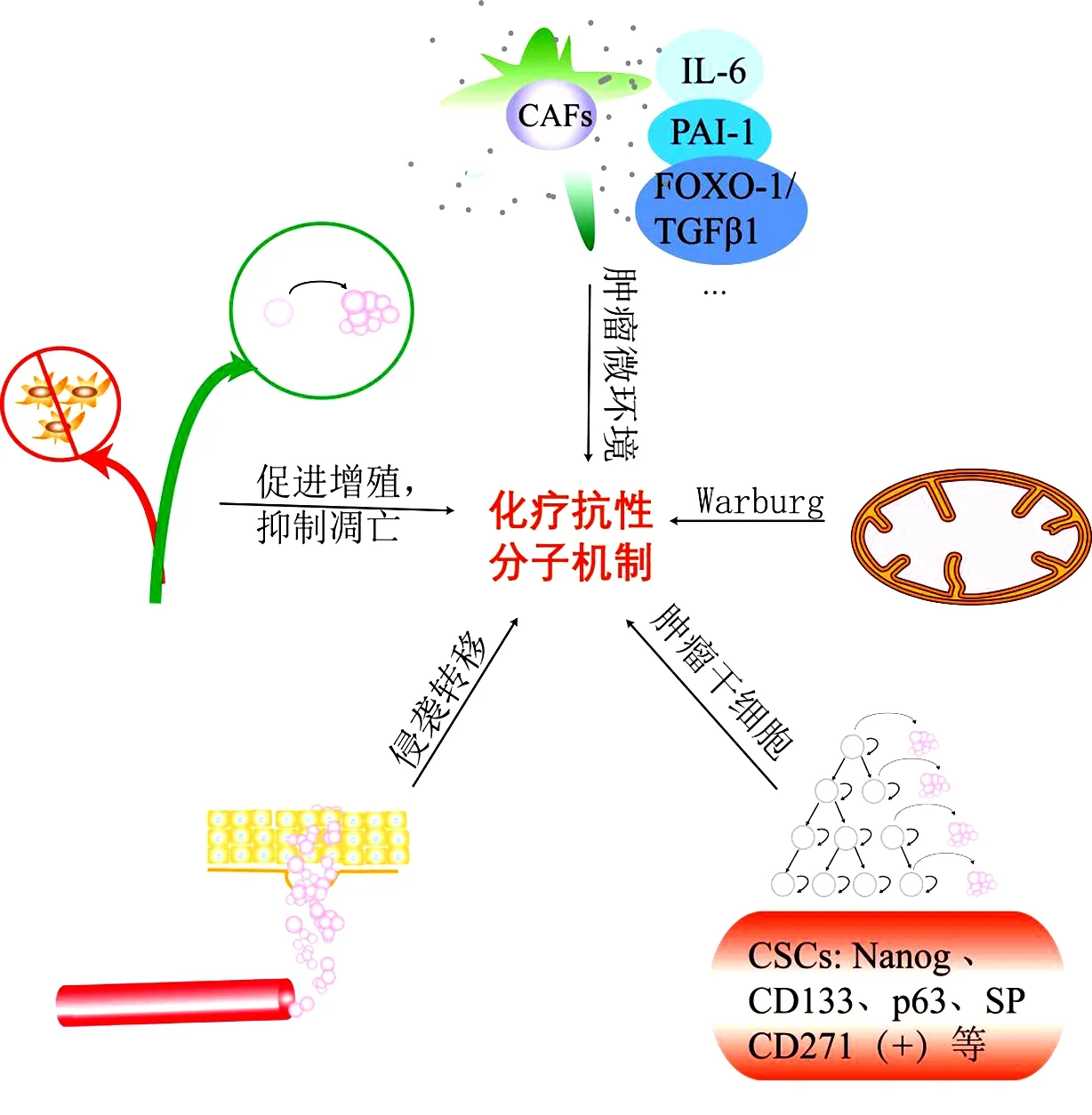
圖3 靶后耐藥模式圖
3.1 抑制細(xì)胞凋亡、促進(jìn)細(xì)胞增殖、介導(dǎo)化療抗性
3.1.1 凋亡抑制蛋白家族介導(dǎo)DDP抗性凋亡抑制蛋白(inhibitor of apoptosis proteins,IAPs)的抗細(xì)胞凋亡能力在化療抗性中發(fā)揮重要作用。Survivin是細(xì)胞凋亡抑制蛋白IAPs家族的成員,能夠抑制半胱天冬蛋白酶,阻斷細(xì)胞死亡[31]。miR-214-3p靶向下調(diào)Survivin和CUG-BP1表達(dá),誘發(fā)ESCC細(xì)胞化療增敏[32]。Caspase激活因子Smac通過(guò)拮抗IAPs,促進(jìn)細(xì)胞凋亡。Smac基因敲除顯著抑制DDP誘導(dǎo)的線(xiàn)粒體膜電位塌陷、Caspase激活和細(xì)胞色素c釋放所引起的細(xì)胞凋亡,導(dǎo)致體內(nèi)外DDP耐藥[33]。XIAP(X chromosome-linked inhibitor of apoptosis)是IAPs家族的另一成員,靶向抑制Caspase 3和Caspase 9。HSP90(heat shock protein 90)抑制劑17-AAG和DDP聯(lián)用,通過(guò)抑制AKT/XIAP,激活Caspase-3,PARP降解,誘導(dǎo)細(xì)胞凋亡,介導(dǎo)DDP的增敏[34]。
3.1.2 PI3K/AKT/mTOR途徑激活介導(dǎo)化療抗性PI3K/AKT/mTOR信號(hào)通路在調(diào)控細(xì)胞生長(zhǎng)、存活和凋亡等過(guò)程中發(fā)揮重要作用。AKT將BAD蛋白136絲氨酸殘基位點(diǎn)磷酸化,降低其與Bcl-2的親和力,導(dǎo)致抵抗細(xì)胞凋亡[35]。PPP2R1B基因編碼的磷酸酶PP2A可以使AKT去磷酸化,增強(qiáng)化療的敏感性,而miR-200C靶向抑制PPP2R1B表達(dá),增強(qiáng)對(duì)DDP的抗性[36]。抑制PI3K/AKT/mTOR信號(hào)通路上游負(fù)性調(diào)控因子PTEN,增強(qiáng)化療抗性。Cyclin B1通過(guò)PTEN/AKT途徑,抑制Bcl-2依賴(lài)的線(xiàn)粒體調(diào)節(jié)的內(nèi)源性死亡途徑,產(chǎn)生抗性,且體外實(shí)驗(yàn)顯示,miR-141-3p直接與PTEN的3’-UTR結(jié)合,抑制細(xì)胞凋亡,促進(jìn)EC9706R細(xì)胞獲得耐藥[37,38]。mTOR介導(dǎo)化療抗性。PP242能抑制mTORC1和mTORC2的活性以及mTORC1依賴(lài)的PI3K/AKT的反饋激活,促進(jìn)DDP誘導(dǎo)的細(xì)胞凋亡[39]。
3.1.3 NF-kB介導(dǎo)化療抗性在腫瘤細(xì)胞中,NF-κB的表達(dá)可能是影響以5-FU為主的化療的關(guān)鍵因素[40]。siRNA抑制NF-κB蛋白表達(dá)可以增強(qiáng)5-FU的抗細(xì)胞增殖作用[41]。p65 的siRNA抑制NF-κB的活化,提高針對(duì)5-FU的裸鼠模型的化療敏感性[42]。然而,RNAi方法作用時(shí)間短,極大地限制了其臨床應(yīng)用。在ESCC和EAC中均發(fā)現(xiàn),姜黃素通過(guò)抑制IkBa磷酸化,抑制NF-kB的活化,下調(diào)Bcl-2和CyclinD1,使細(xì)胞周期阻滯,增加細(xì)胞凋亡率[43-44]。
3.1.4 p38與p53以不同方式影響化療p38蛋白作為MAPK家族中的成員與誘導(dǎo)凋亡相關(guān)。miR-29c通過(guò)直接靶向抑制FBXO31基因表達(dá)和p38蛋白激活,在體內(nèi)外誘導(dǎo)細(xì)胞凋亡,抑制細(xì)胞增殖,逆轉(zhuǎn)5-FU的化療耐藥性;而STAT5A直接與miR-29c啟動(dòng)子結(jié)合抑制其轉(zhuǎn)錄,產(chǎn)生相反的效應(yīng)[45]。與p38δ陽(yáng)性的ESCC相比,p38δ陰性的ESCC對(duì)DDP和5-FU耐受性更強(qiáng)[46]。P53基因在70%的EAC中發(fā)生突變,導(dǎo)致化療耐藥。突變型P53基因的異位表達(dá)使P53基因缺失的細(xì)胞對(duì)APR-246敏感,而P53基因敲除或敲降降低了藥物活性。APR-246作為突變型P53復(fù)活劑在CLX和PDX模型中顯示了強(qiáng)大的抗腫瘤活性,并恢復(fù)了對(duì)DDP/5-FU耐藥異種移植模型的化療敏感性[47]。
3.1.5 其他靶點(diǎn)分子及通路lncRNA CCAT1在體內(nèi)外通過(guò)調(diào)節(jié)miR-143/PLK1/BUBR1信號(hào)軸,促進(jìn)細(xì)胞增殖,增強(qiáng)DDP耐藥性[48]。BMI1和Mel18共同抑制c-Myc,增加細(xì)胞凋亡和細(xì)胞毒性,介導(dǎo)對(duì)DDP的增敏[49]。在補(bǔ)充甲基硒酸的EAC細(xì)胞中,硒結(jié)合蛋白SELENBP1穩(wěn)定過(guò)表達(dá),導(dǎo)致細(xì)胞凋亡,細(xì)胞衰老和DDP細(xì)胞毒性增加[50]。在EAC細(xì)胞及小鼠模型中,Deferasirox和DFO作為鐵螯合劑有效地抑制了細(xì)胞對(duì)鐵的攝取,促進(jìn)細(xì)胞內(nèi)鐵的動(dòng)員,降低了細(xì)胞活力和增殖,增強(qiáng)DDP、5-FU和阿霉素對(duì)細(xì)胞的毒性[51]。
3.2 腫瘤干細(xì)胞介導(dǎo)DDP抗性
腫瘤干細(xì)胞(Cancer stem cells,CSCs)或腫瘤起始細(xì)胞(tumor initiating cells,TICs)在腫瘤的發(fā)生、轉(zhuǎn)移和復(fù)發(fā)中發(fā)揮主導(dǎo)作用,同時(shí)也可能是化療失敗的源頭。①干細(xì)胞標(biāo)志物與耐藥:Nanog蛋白決定胚胎發(fā)育過(guò)程中多能內(nèi)細(xì)胞團(tuán)(inner cell mass,ICM)的命運(yùn),介導(dǎo)DDP抵抗[52]。CD271+、整合素α6bri/CD71dim和/或過(guò)表達(dá)Achaete-Scute復(fù)合物同源物2似乎至少代表一個(gè)干細(xì)胞亞群,抑制對(duì)5-FU和DDP的敏感性[53-54]。②抑制干細(xì)胞分子標(biāo)志物介導(dǎo)化療藥物增敏:IGF2的中和抗體,通過(guò)IGF2-PI3K/AKT-miR-377-CD133信號(hào)軸,抑制腫瘤干性,增強(qiáng)裸鼠移植瘤對(duì)5-FU治療的敏感性[55]。塞來(lái)昔布通過(guò)抑制COX2表達(dá),減少CSC分子標(biāo)志物,使食管癌細(xì)胞對(duì)5-FU敏感[56]。低濃度的全反式維甲酸(all-trans retinoic acid,ATRA)與5-FU和DDP聯(lián)合使用,可促進(jìn)CSCs凋亡,以及G2/M和G0/G1期細(xì)胞周期阻滯,增強(qiáng)DDP和5-FU的細(xì)胞毒作用[57]。③侵襲與耐藥:干細(xì)胞相關(guān)蛋白,Nanog、p63和Bmi-1,以及EMT相關(guān)蛋白,N-cadherin和纖維粘連蛋白,在P75NTR陽(yáng)性組中表達(dá)水平顯著增高,對(duì)DDP的抗性也顯著增高[58]。BMP結(jié)合卵泡抑素樣蛋白FSTL1,通過(guò)典型的NFκB途徑激活和BMP途徑減弱,在ESCC的發(fā)生和轉(zhuǎn)移中發(fā)揮重要的癌蛋白的作用,介導(dǎo)DDP抗性[59]。染料外排亞群(side population,SP)細(xì)胞作為人食管癌干細(xì)胞(esophageal cancer stem cells,ECSCs)樣亞群的一個(gè)關(guān)鍵特征,對(duì)化療的抵抗力顯著高于大多數(shù)其他癌細(xì)胞。長(zhǎng)期應(yīng)用化療可能通過(guò)SP細(xì)胞的富集,使原本對(duì)化療敏感的食管癌產(chǎn)生獲得性耐藥,促進(jìn)癌細(xì)胞向遠(yuǎn)處轉(zhuǎn)移[60]。④miRNAs與耐藥或增敏:如let7、miR-134、miR-296、miR-302、miR-367和miR-470,參與干細(xì)胞功能調(diào)節(jié),如自我更新、多能性和分化。沉默miR-455-3p使包括Wnt/β-catenin和TGF-β等多條TIC相關(guān)通路失活,減少CD90+和CD271+TIC亞群,可使ESCC細(xì)胞對(duì)DDP增敏[61]。
3.3 TME改變調(diào)控DDP抗性
化療耐藥性是一種復(fù)雜的多因素參與的過(guò)程,同癌細(xì)胞與TME之間的相互作用有關(guān)。越來(lái)越多的證據(jù)表明癌細(xì)胞、成纖維細(xì)胞、內(nèi)皮細(xì)胞、巨噬細(xì)胞和ECM成分組成的TME在促進(jìn)實(shí)體腫瘤對(duì)化療耐藥的發(fā)生中發(fā)揮著關(guān)鍵作用。IL-6通過(guò)激活STAT3/NF-κB信號(hào)通路,上調(diào)CXCR7的表達(dá),介導(dǎo)DDP抗性[62]。涉及癌癥相關(guān)成纖維細(xì)胞(cancer-associated fibroblasts,CAF)的化療耐藥機(jī)制包括調(diào)控癌細(xì)胞與ECM相互作用的通路、CAF-ECM黏附和細(xì)胞因子或趨化因子介導(dǎo)的信號(hào)轉(zhuǎn)導(dǎo)。在腫瘤進(jìn)展的早期階段,TGFβ1與其受體的胞外區(qū)結(jié)合抑制癌細(xì)胞的增殖,在晚期促進(jìn)上皮向間充質(zhì)轉(zhuǎn)化和轉(zhuǎn)移。CAF可通過(guò)FOXO-1/TGFβ1信號(hào)環(huán)介導(dǎo)DDP以及紫杉醇耐藥[63]。PAI-1作為DDP刺激CAF的旁分泌因子可以通過(guò)激活A(yù)KT/ERK,影響體外培養(yǎng)的食管癌細(xì)胞增殖和耐藥,并且PAI-1使caspase和γH2AX失活,降低ESCC細(xì)胞的DNA損傷和ROS水平,抑制氧化應(yīng)激,以保護(hù)腫瘤細(xì)胞免受DDP的影響[64]。
3.4 侵襲轉(zhuǎn)移介導(dǎo)DDP抗性
激活后的SOX9通過(guò)與miR-203a啟動(dòng)子結(jié)合抑制其轉(zhuǎn)錄,阻止miR-203a對(duì)PI3K/AKT/mTOR的抑制作用,促進(jìn)ESCC侵襲,介導(dǎo)DDP抗性[65]。miR-125a-5p的過(guò)表達(dá)顯著降低STAT3和p-STAT3及其下游靶基因VEGF在ESCC細(xì)胞的蛋白水平,介導(dǎo)DDP增敏[66]。達(dá)沙替尼通過(guò)抑制PI3K/AKT和STAT3通路,顯著提高DDP對(duì)ESCC細(xì)胞KYSE410的Caspase的激活,促進(jìn)細(xì)胞凋亡,抑制ESCC細(xì)胞侵襲和血管生成[67]。DDK3處理的EAC細(xì)胞OE33可以通過(guò)TGF-β促進(jìn)血管內(nèi)皮形成,對(duì)5-FU和DDP的耐藥性明顯增強(qiáng)[68]。miR-221直接靶向抑制DKK2激活Wnt/β-catenin通路,促進(jìn)EMT,最終導(dǎo)致化療抵抗[69]。
3.5 代謝
在大多數(shù)情況下,癌細(xì)胞主要依賴(lài)有氧糖酵解提供能量和代謝物質(zhì)。實(shí)體腫瘤組織中的缺氧狀態(tài)不僅為ESCC細(xì)胞的生長(zhǎng)與增殖創(chuàng)造了獨(dú)特的環(huán)境,同時(shí)也使ESCC對(duì)包括化療在內(nèi)的常規(guī)癌癥治療的反應(yīng)性減弱。Warburg效應(yīng)調(diào)節(jié)因子,SLC2A1又稱(chēng)GLUT1,是主要的葡萄糖轉(zhuǎn)運(yùn)蛋白之一,可促進(jìn)糖酵解效率增加,以滿(mǎn)足細(xì)胞生長(zhǎng)的需要。突變型P53的表達(dá)可促進(jìn)GLUT1從細(xì)胞質(zhì)到細(xì)胞表面的轉(zhuǎn)位,增加葡萄糖的攝取。PAX5編碼基因敲低可以通過(guò)PAX5-p53-SLC2A1軸促進(jìn)細(xì)胞增殖,增強(qiáng)DDP抗性[70]。AMPK(AMP-activated protein kinase)是Warburg效應(yīng)的負(fù)調(diào)節(jié)因子,其活性形式可下調(diào)能量消耗過(guò)程。水飛薊賓可以激活A(yù)MPK,逆轉(zhuǎn)Warburg表型,協(xié)同5-FU發(fā)揮抗癌作用[71]。ATP調(diào)節(jié)因子,ALC1是ATP依賴(lài)的染色質(zhì)重塑酶,它通過(guò)抑制PI3K/AKT途徑,抑制糖酵解,抑制細(xì)胞生長(zhǎng),增強(qiáng)DDP對(duì)ESCC細(xì)胞的細(xì)胞毒作用[72]。RAC1的沉默抑制AKT/FOXO3a信號(hào)轉(zhuǎn)導(dǎo),抑制細(xì)胞糖酵解,增敏DDP的治療[73]。磷酸戊糖途徑(pentose phosphate pathway,PPP)將葡萄糖代謝與核酸生物合成聯(lián)系起來(lái),提供谷胱甘肽還原酶所需的NADPH,產(chǎn)生用于ROS解毒的GSH。丙酮酸激酶是糖酵解中的限速酶,抑制丙酮酸激酶M2(pyruvate kinase M2,PKM2),減少葡萄糖流入PPP,恢復(fù)對(duì)DDP的敏感性,導(dǎo)致DDP治療后細(xì)胞內(nèi)ROS水平的增加[74]。YQ23作為一種新型的氧氣載體,能抑制HIF-1α的表達(dá),進(jìn)而使腫瘤表型從較多的間質(zhì)狀態(tài)轉(zhuǎn)變?yōu)樯掀顟B(tài),逆轉(zhuǎn)對(duì)DDP和5-FU的抗性[75]。
受限的葡萄糖水平已被證明可以誘導(dǎo)細(xì)胞周期停滯,減緩細(xì)胞增殖速度,使腫瘤細(xì)胞進(jìn)入靜止?fàn)顟B(tài)。在缺糖條件下,糖酵解受限,二甲雙胍處理的EC109細(xì)胞的能量供應(yīng)受到明顯限制,AKT磷酸化受到明顯抑制,嚴(yán)重?fù)p害了細(xì)胞的DNA修復(fù)過(guò)程[76]。二甲雙胍在體內(nèi)外均能顯著抑制4EBP1和S6K1的表達(dá),促進(jìn)細(xì)胞凋亡,增強(qiáng)DDP的作用[77]。
4 總結(jié)
順鉑、5-FU、紫杉醇的作用部位前中后的機(jī)制有相似之處。三者靶前耐藥機(jī)制主要為膜轉(zhuǎn)運(yùn)蛋白ABC家族促進(jìn)藥物外排。靶上耐藥機(jī)制則因藥物本身作用部位不同而異,如順鉑與DNA損傷修復(fù)相關(guān),5-FU與核苷酸代謝相關(guān),紫杉醇則與G2M期相關(guān)。靶后耐藥與增殖、凋亡、腫瘤干細(xì)胞、腫瘤微環(huán)境、侵襲轉(zhuǎn)移以及代謝等相關(guān)。盡管耐藥機(jī)制令人望而生畏,但不應(yīng)忽視這樣一個(gè)事實(shí),即化療在多數(shù)情況下都是有效的,顯著延長(zhǎng)了患者的生命,在某些情況下,還能治愈食管癌。因此,尋找化療耐藥或增敏的關(guān)鍵分子,減少化療耐藥,是現(xiàn)在以及未來(lái)的重點(diǎn)和難點(diǎn)。
猜你喜歡
保健醫(yī)苑(2022年5期)2022-06-10 07:46:38
現(xiàn)代臨床醫(yī)學(xué)(2022年3期)2022-06-06 07:59:40
世界科學(xué)技術(shù)-中醫(yī)藥現(xiàn)代化(2022年2期)2022-05-25 13:17:14
昆明醫(yī)科大學(xué)學(xué)報(bào)(2022年1期)2022-02-28 07:43:40
昆明醫(yī)科大學(xué)學(xué)報(bào)(2021年12期)2021-12-30 07:00:10
科學(xué)大眾(2020年12期)2020-08-13 03:22:22
實(shí)用口腔醫(yī)學(xué)雜志(2017年6期)2017-09-19 02:51:06
中外醫(yī)療(2016年15期)2016-12-01 04:25:50
哈爾濱醫(yī)藥(2015年2期)2015-12-01 03:57:41
中國(guó)當(dāng)代醫(yī)藥(2015年17期)2015-03-01 02:03:58
