基于組基礎模型的HIV /AIDS發病和死亡縱向資料雙軌跡分析*
2021-07-07 09:37:08高洪艷
中國衛生統計 2021年3期
郭 劍 高洪艷 王 媛
【提 要】 目的 介紹組基礎模型雙軌跡分析原理,以HIV/AIDS發病和死亡縱向資料演示。方法 建立HIV/AIDS發病、死亡組基礎模型,確定最優軌跡組數目和發展軌跡形態,將發病、死亡組基礎模型參數帶入雙軌跡分析,以概率形式反映1990-2015年195個國家或地區HIV/AIDS發病、死亡軌跡組間關聯性。結果 組基礎模型將HIV/AIDS發病和死亡都分為4組:高水平、中高水平、中低水平、低水平。雙軌跡分析表明,發病高水平組成為死亡高水平組概率為66.7%,成為死亡中高水平組概率為33.3%;發病中高水平組成為死亡中高水平組概率為80%,成為死亡高水平組概率為20%。發病低水平組、中低水平組與死亡相應組別100%對應。結論 組基礎模型雙軌跡分析能以概率形式反映兩測量結局發展軌跡間關聯程度和離散程度。
組基礎模型雙軌跡分析(group-based model dual trajectory analysis,GBMDTA)被用來分析兩個測量結局縱向資料的關聯性。在縱向研究中,研究者可能關心兩個不同測量指標的關聯程度。同時期的,如成年人血壓與血脂;不同時期的,如兒童期肥胖和成年血壓。傳統用于表述兩個不同但存在關聯的測量指標間關系的統計量主要是相關系數、比值比等。這樣最多只關注兩個時期的統計方法,無法充分利用縱向研究數據信息。而且超過兩個時期的縱向數據能反映的遠不止一條線性發展軌跡,這也是傳統的匯總式統計量無法有效反映的。GBMDTA通過聯接兩個不同指標的發展軌跡,以概率形式評價兩者關聯程度。最早,由Nagin等人用于分析兒童期過渡活躍與成年期焦慮的關系[1],在心理學領域有所應用。而公共衛生領域罕有報道。本研究旨在介紹GBMDTA原理,并以HIV/AIDS發病和死亡縱向資料演示,以推動其在公共衛生領域的應用。
資料與方法
1.資料來源
資料來源于全球疾病負擔研究(global burden of disease,GBD 2017)195個國家或地區1990年、1995年、2000年、2005年、2010年、2015年HIV/AIDS年齡標化發病率和死亡率。
2.基本原理[2]
(1)GBMDTA原理
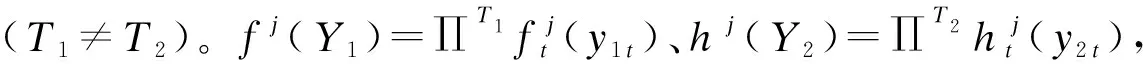
似然函數:假設Y1的J個軌跡組與Y2的K個軌跡組存在概率上的聯系,以j和k,Y1與Y2為條件的分布相互獨立,則Pjk(Y1,Y2)=fj(Y1)hk(Y2)。因此,Y1與Y2的非條件似然函數為每個個體的Pjk(Y1,Y2)合計,并且每個這種條件分布以πjk(屬于Y1的j個軌跡組和Y2的k個軌跡組的成員聯合概率)為權重。
(1)
公式(1)中πjk=πk|jπj,因此公式(1)也可寫作:
(2)
公式(2)中的似然函數由兩個部分按順序組成,Y1的每個j組與Y2的每個k組通過條件概率πk|j聯接。公式(2)自然反映了Y1在時間上先于Y2的順序。然而,無論孰先孰后,公式(2)也有另一種對等形式,通過條件概率πj|k聯接每個k組與每個j組。對于每個個體的似然函數公式可表達為:
(3)
描述兩個結局軌跡間關聯的概率用πj|k、πk|j和πjk表示,說明兩結局發展軌跡的重疊程度。公式(1)~(3)中涉及πj、πk、πj|k,πk|j和πjk,計算過程介紹如下:
①πj表示一個隨機選取的個體Y1屬于某一亞組j的概率,即總體中第j個亞總體的比例:
(4)
其中,θj,j=1,2,…,j,是由不帶協變量的多項式logit模型估計所得的參數。因各組πj相加等于1,只需計算J-1個θj。
②πk表示一個隨機選取的個體Y2屬于某一亞組k的概率。
(5)
πjk=πk|jπj
(6)
公式(5)表示Y2的k個軌跡組中每一個組的成員概率。公式(6)說明公式(5)中每一部分是屬于軌跡組k和j的成員聯合概率,πjk=πk|jπj,即在給定j軌跡組條件下,屬于第k組的概率。為計算πk需要對Y1的j個組的聯合概率求合計。因為個體屬于Y2的某一軌跡組k的同時,也屬于Y1的J個軌跡組中的一組,J個軌跡組的聯合概率πjk的合計等于πk。
③πj|k可用于反映Y1的j軌跡組對Y2特定的軌跡組k的貢獻。即在給定某些k軌跡組時,個體屬于特定j軌跡組的概率。
(7)
④πk|j在給定軌跡組j條件下,個體屬于軌跡組k的概率。
(8)
其中,γk|j,j=1,2,…,J,k=1,2,…,K,是由不帶協變量的多項式logit模型估計所得的參數。
公式(8)需要計算J×K個概率。對每一個Y1的J個軌跡組,都有K個轉換概率,對應Y2的K個軌跡組之一。因此,每個亞組j需要估計K-1個參數,每一個對應Y2的K-1個軌跡組之一。第K組的轉換概率能夠通過1減其他K-1個組的概率得到。因此,公式8需要估計J×(K-1)個參數。即為Y1的J個軌跡組中的每個亞組估計K-1個參數。
GBMDTA的兩個概念模型。在限制模型中Y1的每個軌跡都與Y2的軌跡單獨關聯。研究者可以假設特定Y1與Y2軌跡一一對應。在全模型中Y1與Y2間的關聯限制被去除,取而代之的是采用概率描述軌跡間的關聯。這種多方面關聯允許模型反映軌跡間聯接特征的模式,研究者也不必假定兩個不同結局的關聯形式(圖1)。本研究采用全模型的形式演示GBMDTA分析過程。
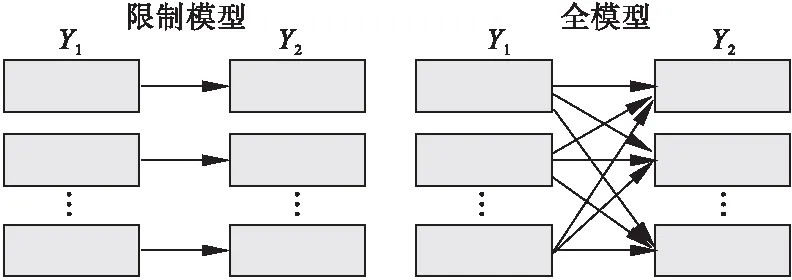
圖1 概念模型
(2)分析步驟
建立單指標組基礎模型,確定最優軌跡組數目和發展軌跡形態,依據BIC(接近0者擬合優度最好)篩選不同模型[3]。確定模型后,將兩個單指標組基礎模型所得參數代入GBMDTA。
(3)統計軟件
采用SAS 9.4統計軟件包的PROC TRAJ過程實施GBMDTA擬合。
結 果
1.單指標組基礎模型擬合
HIV/AIDS年齡別標化發病率和死亡率為正偏態分布資料。因此,本研究將其進行標準化正態變換后再分析。
模型選擇過程:從1組開始逐步增加軌跡組數目,嘗試各軌跡組形態包括:常數、線性、2次曲線、3次曲線。將BIC作為篩選標準選擇最佳模型(BIC越接近0越好),具體如下:
HIV/AIDS發病率:1組 BIC=-1664.22(N=1170),BIC=-1662.42(N=195);2組 BIC=-1023.95(N=1170),BIC=-1016.78(N=195);3組 BIC=-897.68(N=1170),BIC=-888.72(N=195);4組BIC=-892.64(N=1170),BIC=-880.10(N=195)。因分4組時BIC最接近于0,最終確定發病率分為4個軌跡組,從低到高各組軌跡形態分別為:線性、線性、線性、3次曲線。地理分布:高水平組(4.6%)多位于非洲南部;中高水平組(2.7%)多位于非洲東部;中低水平組(6%)多位于非洲中西部;低水平組(86.6%)位于世界各地(圖2)。發病率GBM估計結果見表1。

表1 HIV/AIDS發病率GBM參數估計結果
HIV/AIDS死亡率:1組BIC=-1664.22(N=1170),BIC=-1662.42(N=195);2組BIC=-1093.07(N=1170),BIC=-1086.80(N=195);3組BIC=-940.96(N=1170),BIC=-932.00(N=195);4組BIC=-884.44(N=1170),BIC=-872.79(N=195)。因分4組時BIC最接近于0,最終確定死亡率分為4個軌跡組,從低到高各組軌跡形態分別為:線性、線性、線性、2次曲線。地理分布范圍與發病率相近:高水平組(3.6%)、中高水平組(4.1%)、中低水平組(7.7%)、低水平組(84.7%)(圖2),死亡率GBM估計結果見表2。
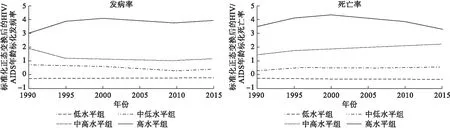
圖2 1990-2015年全球HIV/AIDS發病率、死亡率軌跡分組

表2 HIV/AIDS死亡率GBM參數估計結果
2.HIV/AIDS發病與死亡GBMDTA
我們將表1、表2中發病率、死亡率GBM估計結果所得參數帶入GBMDTA中作為模型參數估計的初始值,擬合模型。
因概率基于發病率軌跡組,表3中每列合計為1。發病率高水平組成為死亡率高水平組可能性為66.7%,成為死亡率中高水平組可能性為33.3%;發病率中高水平組成為死亡率中高水平組概率為80%,成為死亡率高水平組概率20%。發病率低水平組、中低水平組分別與死亡率相應組別100%對應(表3)。
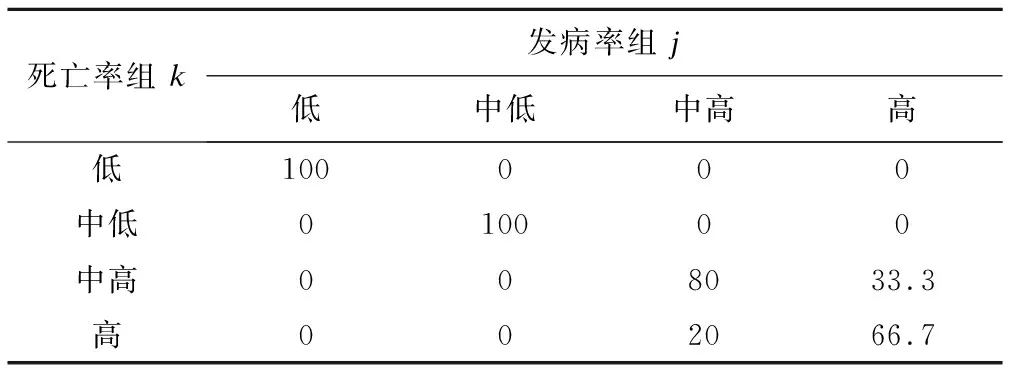
表3 基于發病率組j條件的死亡率組k的概率(πk|j,%)
因概率基于死亡率軌跡組,表4中每行合計為1。死亡率高水平組85.7%歸因于發病率高水平組,14.3%歸因于發病率中高水平組;死亡率中高水平組57.1%歸因于發病率中高水平組,42.9%歸因于發病率高水平組。死亡率低水平組、中低水平組分別100%歸因于發病率的低水平組、中低水平組(表4)。

表4 基于死亡率組k條件的發病率組j的概率(πj|k,%)

①πk=3,4|j=3為表1中發病率中高組對死亡率中高組、高組概率合計80%+20%=100%。
②πj為總體估計比例,前面發病率組基礎模型時4組比例,低水平組πj=1=86.6%,中低水平組πj=2=6%,中高水平組πj=3=2.7%,高水平組πj=4=4.6%。
③πk=3,4可根據公式(5)利用πj和表1中的πk|j求得:
πk=3=πk=3|j=1πj=1+πk=3|j=2πj=2+πk=3|j=3πj=3+πk=3|j=4πj=4=0×86.6%+0×6%+80%×2.7%+33.3%×4.6%=3.7%
同樣可求得:
πk=4=3.6%
πk=3,4=πk=3+πk=4=3.7%+3.6%=7.3%。

因此,死亡率的中高水平組和高水平組37%歸因于發病率的中高水平組。
表5是發病率軌跡與死亡率軌跡的聯合概率,總計4×4=16個聯合概率的合計為1。結果表明,84.9%的國家同時屬于發病率和死亡率的低水平組,7.9%屬于發病率和死亡率中低水平組,3.1%屬于發病率和死亡率高水平組,其余以此類推(表5)。
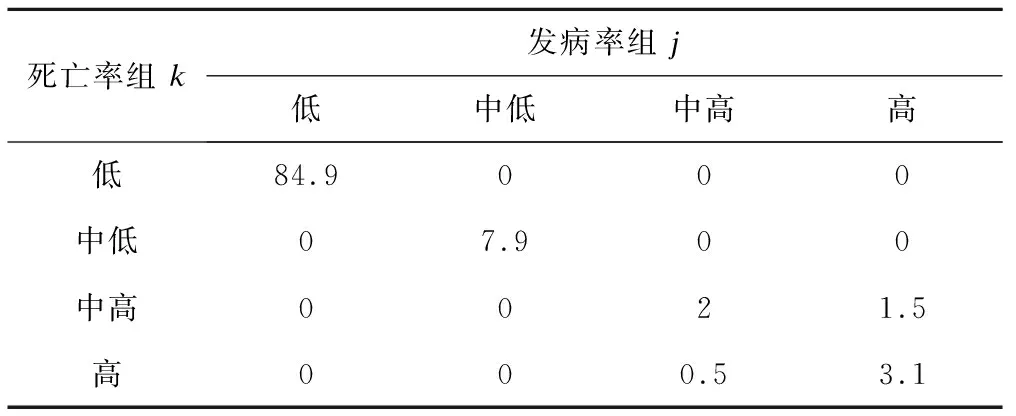
表5 發病率組j和死亡率組k的聯合概率(πjk,%)
3.HIV/AIDS發病率和死亡率地理分布
圖3為全球195個國家發病率、死亡率地理分布圖。HIV/AIDS發病、死亡水平在全球分布并不均衡。以非洲中部、南部最為嚴重,其余各地較低[4]。從中可見HIV/AIDS發病、死亡分布范圍相似,與GBMDTA結果基本一致。

圖3 2015年全球195個國家或地區HIV/AIDS發病率和死亡率
討 論
本研究結果表明,HIV/AIDS發病率、死亡率都被分為4個軌跡組。GBMDTA表明,發病率高水平組成為死亡率高水平組可能性為66.7%,成為死亡率中高水平組可能性為33.3%;發病率中高水平組成為死亡率中高水平組概率為80%,成為死亡率高水平組概率20%。發病率低水平組、中低水平組分別與死亡率相應組別100%對應。大多數國家(84.9%)同時屬于發病率和死亡率低水平組。很少國家(3.1%)的國家同時屬于發病率和死亡率高水平組。
GBMDTA是建立在單指標組基礎模型之上的分析方法,通過事先將測量結局劃分為不同等次的軌跡組描述縱向資料的發展軌跡,區分了資料的異質性。傳統統計方法是假設所有個體來自同一研究總體,即反映異質性的亞總體的平均相關程度。事實上某些亞總體可能只存在較強相關,其他亞總體可能存在微弱關聯。
另一方面,GBMDTA能反映兩個結局測量的軌跡組趨勢,每個軌跡組中的成員概率,跨軌跡組間的關聯成員概率。采用軌跡組的形式總結感興趣結局的發展軌跡以發掘縱向資料的特征,更專注于兩個變量間動態重疊的幅度。相比之下,傳統統計方法,最多只關注兩個時期,僅反映同期、非同期關聯,未能充分利用耗資龐大的縱向研究數據信息。而且,GBMDTA通過總結每種結局的軌跡組的跨組關聯,以概率的形式表達,而非單一的總結性統計量,不僅能反映軌跡組間平均趨勢,還能闡釋對平均趨勢的離散程度,為反映兩個變量間發展過程的內在聯系模式提供了豐富的信息。
GBMDTA通過πj|k,πk|j和πjk三組概率可以描述兩個發展軌跡的關聯程度,可以幫助研究者探索以下問題:Y2特定軌跡組的成員概率是否獨立于Y1的軌跡組成員概率?這有助于評價Y1與Y2發展軌跡的關聯,即兩變量間平均趨勢的關聯程度。本研究中各國家的HIV/AIDS發病率與死亡率軌跡就存在著密切的對應關系。另一方面,通過Y1與Y2聯合成員概率,判定分布于極端軌跡組的亞群大小。這有助于評價針對特定極端亞組開展干預項目的成本效益。本研究中發病率高水平組與死亡率高水平組聯合概率3.1%,主要包含位于非洲南部的7個國家。相比低流行區,這些地區為HIV/AIDS流行的重災區,開展針對性的防控項目成本效益更為合理。
GBMDTA僅限于兩個結局的關系,無法滿足分析兩個以上結局隨時間變化關系的需求。因此,有研究者建議采用結構方程模型思路[5],構建兩個以上結局測量發展過程的線性或非線性潛發展模型(latent growth model with multiple growth process),這一思路與GBMDTA一樣也包括多測量結局同時存在的平行發展過程(parallel growth processes)和具有前因后果的順序發展過程(sequential growth process)[6-7]。然而,隨著測量結局數量增多,結構方程模型復雜性也升高,要估計的參數也增加,模型所需樣本量也增大。
GBMDTA能在區分總體異質性的前提下探索兩個結局各軌跡亞組間的關聯性。在公共衛生領域中,可用于縱向研究探索兩研究因素的關聯程度,如一定時期內某地高危性行為頻率與HIV/AIDS流行情況關系,或個體腰臀比變化對血壓影響等。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:30
當代陜西(2021年17期)2021-11-06 03:21:36
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
當代陜西(2019年15期)2019-09-02 01:52:00
學苑創造·A版(2018年11期)2018-02-01 06:29:20
讀者(2017年5期)2017-02-15 18:04:18
光學精密工程(2016年6期)2016-11-07 09:07:19
核科學與工程(2015年4期)2015-09-26 11:59:03
