不同炮制時間的荊芥炭色度值與多項指標的相關性研究
2021-07-12 09:13:26田甜彭幫貴徐杰洪婉敏劉路芳何海兵魏梅
中國藥房 2021年12期
田甜 彭幫貴 徐杰 洪婉敏 劉路芳 何海兵 魏梅


摘 要 目的:研究不同炮制時間(0~40 min,下同)的荊芥炭色度值與多項指標的相關性,揭示荊芥炭在炮制過程中的質量變化規律,確定炮制終點時間。方法:測定不同炮制時間的荊芥炭飲片中醇溶性浸出物的含量;建立荊芥飲片及不同炮制時間荊芥炭飲片的超高效液相色譜(UPLC)指紋圖譜并進行相似度評價,通過與對照品比對確認色譜峰;采用相同的UPLC條件測定不同炮制時間荊芥炭飲片中指標成分(橙皮苷、迷迭香酸、胡薄荷酮)的含量;運用色差儀測定不同炮制時間荊芥炭飲片的色度值,以炮制0 min樣品為對照,計算總色值(E)和總色差值(ΔE)。對荊芥炭飲片中醇溶性浸出物、指標成分含量及色譜峰峰面積與色度值參數進行Pearson相關性分析、聚類分析和正交偏最小二乘法判別分析。確定荊芥炭炮制終點時間并驗證。結果:隨著炮制時間的延長,荊芥炭飲片中醇溶性浸出物的含量逐漸降低。12批荊芥飲片中共標定17個色譜峰,與對照指紋圖譜的相似度均大于0.9。不同炮制時間荊芥炭飲片中共標定21個色譜峰,與炮制0 min樣品圖譜的相似度隨著炮制時間延長而降低,其中炮制18 min后的相似度均低于0.9。確認色譜峰峰9為橙皮苷、峰10為迷迭香酸、峰17為胡薄荷酮。含量和色度值測定結果顯示,隨著炮制時間的延長,橙皮苷、迷迭香酸、胡薄荷酮含量均逐漸降低,荊芥炭飲片粉末顏色L、b、E值均逐漸減小,a、ΔE值均逐漸增大。Pearson相關性分析顯示,荊芥炭飲片中醇溶性浸出物含量、橙皮苷含量、迷迭香酸含量、胡薄荷酮含量以及15個色譜峰(峰2、7~15、17~21)的峰面積與E值成顯著正相關(P<0.01),5個色譜峰(峰1、3~6)的峰面積與E值成顯著負相關(P<0.01),而峰16的峰面積與E值無關(P>0.05)。聚類分析結果顯示,不同炮制時間的荊芥炭飲片被分成2類,炮制0~16 min為第1類、18~40 min為第2類。正交偏最小二乘法判別分析結果顯示,荊芥炭飲片的峰6峰面積(2.800 75)、L值(2.327 54)、峰3峰面積(1.793 39)、b值(1.735 78)、峰5峰面積(1.244 04)的變量投影重要性(VIP值)均大于1。荊芥炭炮制終點時間為18 min;3次驗證實驗結果顯示,荊芥炭飲片的L、a、b值分別為20.22~22.00、4.44~7.67、9.78~13.00,ΔE為13.50~14.12。結論:不同炮制時間荊芥炭飲片的色度值與其醇溶性浸出物含量、橙皮苷含量、迷迭香酸含量、胡薄荷酮含量以及20個色譜峰的峰面積密切相關。建議荊芥炭炮制終點時間為18 min。
關鍵詞 荊芥;荊芥炭;炮制時間;超高效液相色譜法;指紋圖譜;色度值;含量;相關性
ABSTRACT ? OBJECTIVE: To study the correlation of the chromaticity value of Schizonepeta tenuifolia charcoal of different processing time (0-40 min, similarly hereinafter) with multiple indicators, and to reveal the quality change law of S. tenuifolia charcoal during processing and confirm the terminal time. METHODS: The contents of ethanol-soluble extracts from S. tenuifolia charcoal decoction pieces of different processing time were determined. UPLC fingerprint of S. tenuifolia decoction pieces and S. tenuifolia charcoal decoction pieces of different processing time were established, and the similarity evaluation was also conducted. The chromatographic peaks were confirmed by comparison with substance control. The same UPLC conditions were used to determine the contents of index components (hesperidin, rosmarinic acid, menthone) in S. tenuifolia charcoal decoction pieces of different processing time. The colorimetric method was used to measure the chromaticity value of S. tenuifolia charcoal decoction pieces of different processing time. Meanwhile, sample of processing 0 min was used as a control to calculate the total color value (E) and the total color difference value (ΔE). ?Pearson correlation analysis, cluster analysis and orthogonal partial least squares discriminant analysis (OPLS-DA) were performed on the ethanol-soluble extracts, index component contents, chromatographic peak area and chromaticity value. The terminal time of processing S. tenuifolia charcoal was confirmed, and validation test was also conducted. RESULTS: With the extension of processing time, the content of ethanol-soluble extract in S. tenuifolia charcoal decoction pieces gradually decreased. A total of 17 chromato- graphic peaks were identified in 12 batches of S. tenuifolia decoction piece, and its similarity with the control fingerprint was greater than 0.9. 21 chromatographic peaks were identified in S. tenuifolia charcoal decoction pieces of different processing time, and its similarity with the chromatogram of sample of processing 0 min decreased with the processing time, and the similarity after 18 min was lower than 0.9. The chromatographic peak 9 was hesperidin, peak 10 was rosmarinic acid and peak 17 was menthone. The determination of content and chromaticity value showed that with the extension of processing time, the contents of hesperidin, rosmarinic acid and menthone decreased gradually; the color L, b and E values of S. tenuifolia charcoal decoction piece powder decreased gradually, and the a and ΔE values increased gradually. Pearson correlation analysis showed that the contents of ethanol-soluble extract, hesperidin, rosmarinic acid and menthone, the peak areas of 15 chromatographic peaks (peak 2, 7-15, 17-21) were significantly positively correlated with E value (P<0.01); the peak areas of 5 chromatographic peaks (peak 1, 3-6) were significantly negatively correlated with E value (P<0.01), but peak area of peak 16 was not related to E value (P>0.05). Results of cluster analysis showed that S. tenuifolia charcoal decoction pieces of different processing time were divided into 2 categories; the first category was processed for 0-16 min, and the second category was processed for 18-40 min. The results of OPLS-DA showed that the VIP values of peak 6 area (2.800 75), L value (2.327 54), peak 3 area (1.793 39), b value (1.735 78) and peak 5 area (1.244 04) were greater than 1. The final processing time of S. tenuifolia charcoal was 18 min. The results of validation experiment showed that the L, a and b values of S. tenuifolia charcoal decoction piece were 20.22-22.00, 4.44-7.67, 9.78-13.00, and ΔE were 13.50-14.12, respectively. CONCLUSIONS: The chromaticity value of S. tenuifolia charcoal decoction pieces of different processing time is closely related to the contents of ethanol-soluble extract, hesperidin, rosmarinic acid, menthone and the area of 20 chromatographic peaks. It is suggested that the terminal time of processing S. tenuifolia is 18 min.
KEYWORDS ? Schizonepeta tenuifolia; Schizonepeta tenuifolia charcoal; Processing time; UPLC; Fingerprint; Chromaticity value; Content; Correlation
荊芥始載于《神農本草經》,別名假蘇,為唇形科植物荊芥Schizonepeta tenuifolia Briq.的干燥地上部分。該藥多在夏、秋二季花開到頂、穗綠時采割,除去雜質,曬干。其味辛、性微溫,歸肺、肝經,具有解表散風、透疹、消瘡的功效,可用于治療感冒、頭痛、麻疹、風疹、瘡瘍初起等病癥[1]。荊芥主要含有揮發油類、黃酮及其苷類、萜類、酚類、甾醇類等化學成分,具有抗病毒、抗炎鎮痛、抗腫瘤、免疫調節、抗菌、止血等藥理作用,是傳統中藥材之一[2]。
荊芥經炒炭后,辛散作用減弱,專于收斂止血,常用于便血、崩漏、產后血暈的各種出血證[3]。2020年版《中國藥典》(一部)收載了荊芥、荊芥炭兩種飲片,并規定荊芥炭飲片的炮制方法如下:取荊芥段,照炒炭法(2020年版《中國藥典》(四部)通則0213)炒至表面焦黑色,內部焦黃色,噴淋清水少許,熄滅火星,取出,晾干[1,4]。傳統的外觀控制認為飲片表面焦黑色、內部焦黃色即符合荊芥炒炭標準[5]。這種感官評判標準主觀性太強,缺少客觀數據,容易導致炮制程度不一、荊芥炭飲片的質量難以有效控制。
已有研究表明,色度值可為中藥飲片質量評價和炮制工藝評價提供技術支持和參考,現已被廣泛應用于中藥領域[6-8]。本研究通過測定荊芥炭炮制過程中醇溶性浸出物含量、指紋圖譜、指標成分(橙皮苷、迷迭香酸、胡薄荷酮)含量和粉末的色度值,結合多元統計學分析方法對色度值與多項測定指標的相關性進行分析,揭示荊芥炭在炮制過程中的質量變化規律,為快速評價其質量提供依據。
1 材料
1.1 主要儀器
本研究所用主要儀器包括HP-C220型精密色差儀(深圳漢普光彩科技有限公司),UPLC H-Class型超高效液相色譜(UPLC)儀(美國Waters公司),C21-WK2102型多功能電磁爐(美的集團有限公司),111B型二兩裝高速中藥粉碎機(浙江瑞安市永歷制藥機械有限公司),ME204E型萬分之一天平、XP26型百萬分之一天平(梅特勒-托利多儀器有限公司),HWS28型電熱恒溫水浴鍋(上海一恒科技有限公司),Milli-Q-Direct超純水系統[默克化工技術(上海)有限公司],KQ-700DE型數控超聲波清洗器(昆山市超聲儀器有限公司),DHG-9147A型電熱恒溫干燥箱(上海精宏實驗設備有限公司)等。
1.2 主要藥品與試劑
橙皮苷(批號110721-201818,純度96.2%)、迷迭香酸(批號111871-201706,純度90.5%)、胡薄荷酮(批號111706-201907,純度99.8%)對照品均購自中國食品藥品檢定研究院;甲醇(分析純)購自西隴科學股份有限公司;乙醇(分析純)購自天津富宇精細化工有限公司;乙腈(色譜純)購自默克化工技術(上海)有限公司;磷酸(色譜純)購自天津市科密歐化學試劑有限公司;其余試劑均為分析純或實驗室常用規格,水為超純水。
12批荊芥飲片由廣東一方制有限公司提供,經廣東一方制藥有限公司魏梅主任藥師和廣州中醫藥大學第二中醫院孫冬梅教授鑒定均為唇形科植物荊芥S. tenuifolia Briq.的干燥地上部分,其來源信息見表1。
2 方法與結果
2.1 荊芥炭飲片樣品的制備
取荊芥飲片(編號S8),按照2020年版《中國藥典》(四部)通則0213項下的荊芥炭炮制方法規定[4],置熱鍋內,用武火炒制40 min,從炮制0 min起,每隔2 min進行取樣,同時用紅外測溫儀測定出鍋溫度,放涼,即得荊芥炭飲片(編號JT00~JT20)。
2.2 醇溶性浸出物測定
按照2020年版《中國藥典》(四部)通則2201項下的熱浸法測定荊芥炭飲片(編號JT00~JT20)中的浸出物[4],溶劑為70%乙醇。結果,隨著炮制時間的延長,荊芥炭飲片的醇溶性浸出物含量逐漸減少,詳見表2。
2.3 指紋圖譜的建立
2.3.1 色譜條件 以Waters CORTECS UPLC T3(150 mm×2.1 mm,1.6 μm)為色譜柱,以乙腈(A)-0.1%磷酸溶液(B)為流動相進行梯度洗脫(0~6 min,10%A;6~11 min,10%A~13%A;11~14 min,13%A~15%A;14~27 min,15%A~20%A;27~46 min,20%A~30%A;46~56 min,30%A~50%A;56~66 min,50%A~60%A),流速為0.30 mL/min,檢測波長為235 nm,柱溫為30 ℃,進樣量為2 ?L。
2.3.2 單一對照品溶液的制備 取橙皮苷、迷迭香酸、胡薄荷酮對照品各適量,精密稱定,加甲醇分別制成每1 mL含橙皮苷61.361 ?g、迷迭香酸31.675 ?g、胡薄荷酮61.756 ?g的單一對照品溶液,備用。
2.3.3 供試品溶液的制備 取荊芥飲片粉末(過二號篩,下同)0.5 g,精密稱定,置于具塞錐形瓶中,精密加入70%甲醇25 mL,密塞,稱定質量,超聲(功率250 W,頻率40 kHz)處理30 min,取出,放冷,再次稱定質量,用70%甲醇補足減失的質量,搖勻,經0.22 μm微孔濾膜濾過,取續濾液,即得。
2.3.4 精密度試驗 精密吸取“2.3.3”項下同一供試品溶液(編號S1)適量,按“2.3.1”項下色譜條件連續進樣測定6次,以橙皮苷色譜峰為參照,計算各色譜峰的相對保留時間和相對峰面積。結果,各色譜峰相對保留時間的RSD均小于0.5%,相對峰面積的RSD均小于3.0%(n=6),表明該方法精密度良好。
2.3.5 重復性試驗 按“2.3.3”項下方法平行制備供試品溶液(編號S1)6份,按“2.3.1”項下色譜條件進樣測定,以橙皮苷色譜峰為參照,計算各色譜峰的相對保留時間和相對峰面積。結果,各色譜峰相對保留時間的RSD均小于1.0%,相對峰面積的RSD均小于3.0%(n=6),表明該方法重復性較好。
2.3.6 穩定性試驗 精密吸取“2.3.3”項下同一供試品溶液(編號S1)適量,分別于室溫下放置0、2、4、8、12、24 h時按“2.3.1”項下色譜條件進樣測定,以橙皮苷色譜峰為參照,計算各色譜峰的相對保留時間和相對峰面積。結果,各色譜峰相對保留時間的RSD均小于1.0%,相對峰面積的RSD均小于3.5%(n=6),表明供試品溶液在室溫下放置24 h內穩定性良好。
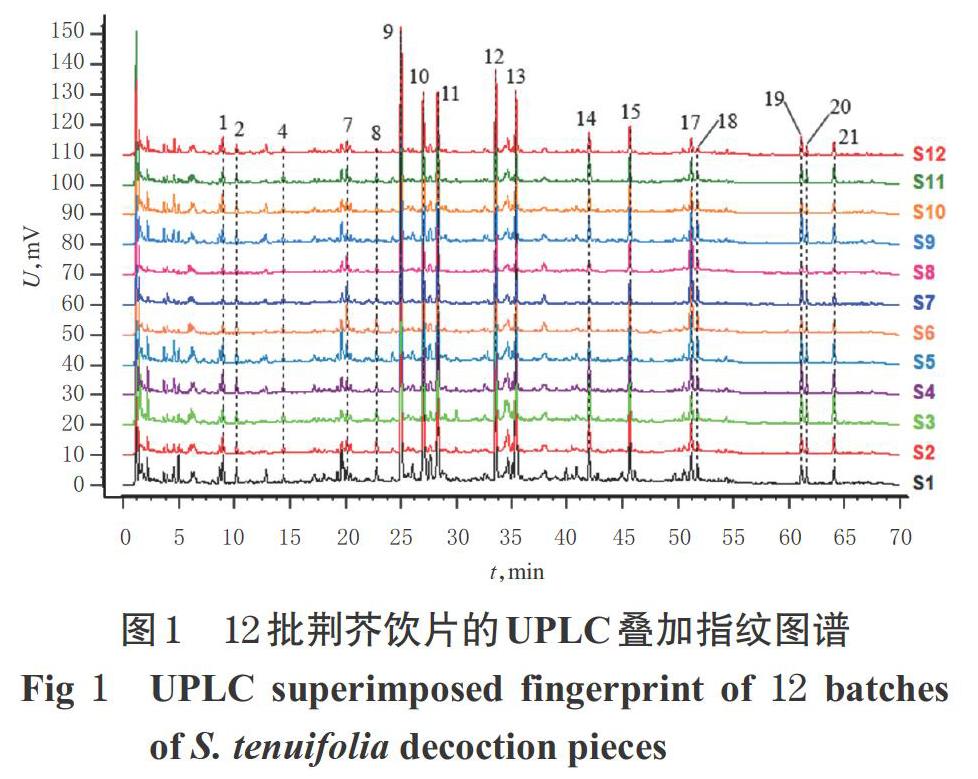
2.3.7 指紋圖譜的建立和色譜峰的指認 取荊芥飲片(編號S1~S12)和不同炮制時間的荊芥炭飲片(編號JT00~JT20),按“2.3.3”項下方法制備供試品溶液,再按“2.3.1”項下色譜條件進樣測定,記錄色譜圖。將色譜圖分別導入《中藥色譜指紋圖譜相似度評價系統(2012A版)》,以編號S1樣品和編號JT00樣品的色譜圖為參照圖譜,將時間窗寬度設為0.1 min,采用多點校正法進行峰匹配,建立荊芥飲片和不同炮制時間的荊芥炭飲片的UPLC疊加指紋圖譜,采用中位數法生成對照指紋圖譜(R)。結果,12批荊芥飲片共標定17個色譜峰,不同炮制時間的荊芥炭飲片共標定21個色譜峰。與對照品(“2.3.2”項下單一對照品溶液以“2.3.1”項下色譜條件測得)比對,確定峰9為橙皮苷、峰10為迷迭香酸、峰17為胡薄荷酮,詳見圖1~圖3。
2.3.8 荊芥飲片的相似度評價 以荊芥飲片的對照指紋圖譜為對照,利用《中藥色譜指紋圖譜相似度評價系統(2012A版)》對12批荊芥飲片的指紋圖譜進行整體相似度評價。結果,12批荊芥飲片指紋圖譜的相似度依次為0.902、0.993、0.980、0.993、0.967、0.952、0.916、0.914、0.966、0.990、0.985、0.995,均大于0.9,表明12批荊芥飲片與對照指紋圖譜具有較好的一致性,飲片批間差異較小。
2.3.9 荊芥炭飲片的相似度評價 以編號JT00樣品圖譜為對照,利用《中藥色譜指紋圖譜相似度評價系統(2012A版)》對不同炮制時間的荊芥炭飲片(編號JT01~JT20)進行相似度評價。結果,隨著炮制時間的延長,荊芥炭飲片指紋圖譜與編號JT00樣品圖譜的相似度逐漸降低;當炮制18 min后,兩者的相似度低于0.9;至炮制40 min時,相似度僅為0.396,詳見表3。
2.3.10 炮制時間對荊芥炭飲片色譜峰的影響 對比圖1和圖2可知,12批荊芥飲片中均未能檢測到峰3、5~6、16,但經炮制后峰3、5~6、16均逐漸被檢測到,說明此4個色譜峰可能為荊芥炒炭后的新增成分,可作為區分荊芥飲片與荊芥炭飲片的關鍵指標。通過比較不同炮制時間荊芥炭飲片色譜峰的峰面積發現,隨著炮制時間的延長,峰1、3~6的峰面積逐漸增加,峰2、14~16的峰面積先增加后減小,其余12個峰的峰面積逐漸減小。
2.4 含量測定
2.4.1 色譜條件 同“2.3.1”項。
2.4.2 單一對照品溶液的制備 同“2.3.2”項。
2.4.3 供試品溶液的制備 同“2.3.3”項。
2.4.4 標準曲線的繪制 分別精密吸取“2.4.2”項下單一對照品溶液0.5、1、2、4、6、8 mL,置于25 mL量瓶中,加甲醇稀釋至刻度,混勻,得系列混合對照品溶液。分別吸取上述混合對照品溶液適量,按“2.4.1”項下色譜條件進樣測定,以各成分的質量濃度為橫坐標(x,?g/mL)、對應峰面積為縱坐標(y)進行線性回歸。結果,橙皮苷、迷迭香酸、胡薄荷酮的回歸方程分別為y=11 100.0x-1 958.9、y=4 866.8x-2 375.7、y=15 794.0x-57 036.0,R 2分別為0.999 2、0.999 5、0.999 9,表明橙皮苷、迷迭香酸、胡薄荷酮質量濃度分別在1.227~19.636、0.634~10.136、1.235~19.762 ?g/mL范圍內與峰面積線性關系良好。
2.4.5 精密度試驗 精密吸取“2.4.4”項下混合對照品溶液(橙皮苷、迷迭香酸、胡薄荷酮質量濃度分別為9.818、5.068、9.881 ?g/mL)適量,按“2.4.1”項下色譜條件連續進樣6次,記錄峰面積。結果,橙皮苷、迷迭香酸、胡薄荷酮峰面積的RSD分別為0.77%、0.89%、2.40%(n=6),表明儀器精密度良好。
2.4.6 穩定性試驗 精密吸取“2.4.3”項下同一供試品溶液(編號S1)適量,分別于室溫下放置0、2、4、8、12、24 h時按“2.4.1”項下色譜條件進樣測定,記錄峰面積。結果,橙皮苷、迷迭香酸、胡薄荷酮峰面積的RSD分別為0.94%、0.70%、3.20%(n=6),表明供試品溶液在室溫下放置24 h內穩定性良好。
2.4.7 重復性試驗 按“2.4.3”項下方法平行制備供試品溶液(編號S1)6份,按“2.4.1”項下色譜條件進樣測定,記錄峰面積。結果,橙皮苷、迷迭香酸、胡薄荷酮含量的RSD分別為0.63%、0.79%、0.81%(n=6),表明該方法重復性較好。
2.4.8 加樣回收率試驗 稱取已知含量的荊芥飲片粉末(編號S1)9份,每份0.5 g,精密稱定,分別按對照品加入量與所取供試品中待測成分量之比0.5 ∶ 1、1 ∶ 1、1.5 ∶ 1精密加入橙皮苷、迷迭香酸、胡薄荷酮對照品(各比例均平行3次),按“2.4.3”項下方法制備供試品溶液,再按“2.4.1”項下色譜條件進樣測定,記錄峰面積并計算加樣回收率。結果,橙皮苷、迷迭香酸、胡薄荷酮的平均加樣回收率分別為95.88%、96.79%、95.65%,RSD分別為1.15%、0.58%、1.54%(n=9),表明該方法回收率良好。
2.4.9 樣品含量測定 取不同炮制時間的荊芥炭飲片(編號JT00~JT20)粉末,按“2.4.3”項下方法制備供試品溶液,再按“2.4.1”項下色譜條件進樣測定,記錄峰面積并代入回歸方程計算橙皮苷、迷迭香酸、胡薄荷酮的含量。結果,隨著炮制時間的延長,橙皮苷、迷迭香酸、胡薄荷酮含量均逐漸降低,詳見表4。
2.5 色度值的測定
2.5.1 測定方法 取不同炮制時間的荊芥炭飲片(編號JT00~JT20)粉末(過三號篩,下同),作為供試品。采用精密色差儀測定色度值:黑白校正后將供試品分別壓制于載玻片上,壓制厚度約為1 mm;測定條件為D65光源,8°視角,孔徑8 mm,LED藍光激發,儀器誤差值小于0.4,記錄供試品的色度值(L、a、b值代表物體顏色的色度值,其中L值越大表示顏色越亮/淺,a值越大表示顏色越偏紅,b值越大表示顏色越偏黃[9])。各樣品平行測定3次,取平均值。
2.5.2 精密度試驗 取荊芥炭飲片(編號JT01)粉末,按“2.5.1”項下方法連續測定6次,記錄色度值。結果,其L、a、b值的RSD均小于3.0%(n=6),表明該方法精密度良好。
2.5.3 重復性試驗 取荊芥炭飲片(編號JT01)粉末6份,按“2.5.1”項下方法測定,記錄色度值。結果,其L、a、b值的RSD均小于2.5%(n=6),表明該方法重復性良好。
2.5.4 穩定性試驗 取荊芥炭飲片(編號JT01)粉末,按“2.5.1”項下方法分別壓制于載玻片上,在室溫下放置0、2、4、6、8、10、12、24 h時測定色度值。結果,其L、a、b值的RSD均小于2.0%(n=8),表明樣品在室溫下放置24 h內色度值的穩定性良好。
2.5.5 總色值和總色差值計算 取不同炮制時間的荊芥炭飲片(編號JT00~JT20)粉末,按“2.5.1”項下方法測定,記錄色度值并計算各樣品的總色值[E=(L2+a2+b2)1/2]、總色差值[ΔE,與編號JT00樣品比較,ΔL=L樣品-L0,Δa=a樣品-a0,Δb=b樣品-b0,ΔE=(ΔL2+Δa2+Δb2)1/2;式中,L0、a0、b0均為編號JT00樣品的色差值][9-10],結果見表5。
由表5可知,荊芥炭飲片在炮制過程中E值隨炮制時間的延長而逐漸減小,主要與L、b值明顯減小有關。L、b值逐漸減小,表明炮制過程中飲片粉末的明亮度由明變暗,藍色加深;a值逐漸增大,表明炮制過程中飲片粉末紅色加深。荊芥炭飲片在炮制過程中ΔE值隨炮制時間的延長而逐漸增大。按照CIELAB均勻色空間系統理論,當ΔE相差6~12個色差單位時,其色差可被人眼識別[9-11];當ΔE≥12時,荊芥炭飲片的外觀顏色符合《中國藥典》相關要求[1]。當炮制18 min后,荊芥炭飲片的ΔE值均大于12;且隨著炮制時間的延長,荊芥炭飲片的ΔE值進一步增大。
2.6 炮制過程中荊芥炭顏色與成分動態關聯分析
2.6.1 Pearson相關性分析 利用SPSS 20.0軟件對不同炮制時間的荊芥炭飲片(編號JT00~JT20)中醇溶性浸出物含量、指標成分含量、色譜峰峰面積與各色度值參數進行Pearson相關性分析,結果見表6。
由表6可見,荊芥炭飲片中醇溶性浸出物含量、橙皮苷含量、迷迭香酸含量、胡薄荷酮含量以及15個色譜峰(峰2、7~15、17~21)的峰面積與E值成顯著正相關(P<0.01),5個色譜峰(峰1、3~6)的峰面積與E值成顯著負相關(P<0.01),而峰16的峰面積與E值無關(P>0.05)。
2.6.2 聚類分析和偏正交最小二乘法判別分析 將不同炮制時間的荊芥炭飲片(編號JT00~JT20)的色度值參數、醇溶性浸出物含量、指標成分含量及色譜峰峰面積導入SIMCA 14.1軟件進行聚類分析和正交偏最小二乘法判別分析(OPLS-DA),從而預測影響荊芥炭飲片中成分變化的重要變量。聚類分析結果顯示,不同炮制時間的荊芥炭飲片被分成了2類,其中炮制時間0~16 min的聚為第1類、18~40 min的聚為第2類,詳見圖4。OPLS-DA結果顯示,模型參數累積貢獻率(R 2)為0.949,預測優度系數(Q 2)為0.898,均大于0.5,表明此模型為優質模型,可用于變量的預測;通過此模型預測得到5個變量投影重要性(VIP值)均大于1的變量,分別是峰6峰面積(2.800 75)、L值(2.327 54)、峰3峰面積(1.793 39)、b值(1.735 78)、峰5峰面積(1.244 04),詳見圖5。上述5個變量的變化程度較為明顯,其中通過L、b值可以直觀判斷荊芥炭飲片的炮制終點。
2.7 荊芥炭炮制的終點時間確定與驗證
在確保荊芥炭飲片符合《中國藥典》相關外觀性狀要求的前提下[1],為避免因炮制時間過長而導致的橙皮苷、迷迭香酸、胡薄荷酮含量下降,故本研究選擇荊芥炭飲片炮制終點時間為18 min。按照炮制時間為18 min的炮制工藝對3批荊芥飲片(編號S3~S5)進行炮制,按“2.5.1”項下方法測定其色度值并計算ΔE值。結果,經過18 min炮制后,所得荊芥炭飲片的L、a、b值分別為20.22~22.00、4.44~7.67、9.78~13.00,ΔE為13.50~14.12,詳見表7。
3 討論
顏色和表觀性狀是中藥材及飲片質量評價的重要指標之一。傳統上,中藥炮制過程及炮制終點的色澤評價方法主要是經驗鑒別及直觀判斷,容易受主觀個體差異和不敏感性的影響,造成產品質量的差異[9-13]。由于色度值能客觀、準確地體現荊芥炭炮制過程中的顏色動態變化,因此本研究以不同炮制時間的荊芥炭飲片粉末為對象,運用精密色差儀對其表觀顏色進行量化,結果更具客觀性。由色度值分析結果可知,隨炮制時間的延長,荊芥炭飲片粉末的顏色逐漸加深,L、b、E值均逐漸減小,a、ΔE值均逐漸增大,粉末顏色由黃綠色逐漸變成焦黑色,炮制程度可被肉眼識別,與傳統性狀評價標準較為一致[13]。可見,將主觀化經驗判定的顏色描述轉化為客觀的檢測數據,可對荊芥炭炮制過程中的質量變化進行實時動態監測,為荊芥炭炮制過程的質量控制及快速評價提供新的手段。
本研究結合色度值、UPLC指紋圖譜、浸出物及多指標含量等對不同炮制時間的荊芥炭飲片進行了多元統計分析。通過比較荊芥飲片及不同炮制時間的荊芥炭飲片的指紋圖譜,發現荊芥經過炒炭,UPLC指紋圖譜存在較大差異。荊芥飲片的UPLC指紋圖譜未能檢測到峰3、5~6、16,而經炮制后上述成分被檢出,推測峰3、5~6、16可能為炮制后荊芥炭新產生的化合物。隨著炮制時間的延長,荊芥炭飲片中橙皮苷、迷迭香酸、胡薄荷酮等12個色譜峰的峰面積較荊芥飲片逐漸減小,表明荊芥飲片所含化學成分在炒炭后發生了變化。荊芥飲片經過炒炭后,辛散作用減弱,產生止血作用,因此猜測峰3、5~6、16這4種新產生的化合物可能與止血的藥效密切相關[13],后續擬進一步驗證。
Pearson相關性結果顯示,醇溶性浸出物含量、橙皮苷含量、迷迭香酸含量、胡薄荷酮含量以及15個色譜峰(峰2、7~15、17~21)的峰面積與E值成顯著正相關,5個色譜峰(峰1、3~6)的峰面積與E值成顯著負相關,說明荊芥炭飲片的成分與其粉末色度值具有緊密相關性。OPLS-DA結果顯示,L、b值的VIP值均大于1,a值的VIP值接近1,說明L、a、b值是荊芥炭炮制過程中的重要變量,通過測定色度值可以預測荊芥炭飲片的炮制程度,以實現對荊芥炭炮制質量的控制。本研究結果顯示,炮制18 min以上的荊芥炭飲片的ΔE均大于12,且隨炮制時間的延長,橙皮苷、迷迭香酸、胡薄荷酮的含量逐漸減少,綜合考慮確定炮制終點為18 min。然而,顏色與藥效水平的關系還需進一步的實驗驗證,以期為荊芥炭飲片質量評價提供更全面的數據支撐。
綜上所述,荊芥炭飲片粉末的色度值與其醇溶性浸出物含量、橙皮苷含量、迷迭香酸含量、胡薄荷酮含量以及20個色譜峰的峰面積密切相關。建議荊芥炭飲片炮制終點時間為18 min。
參考文獻
[ 1 ] 國家藥典委員會.中華人民共和國藥典:一部[S]. 2020年版.北京:中國醫藥科技出版社,2020:243.
[ 2 ] 劉英男,牛鳳菊,辛義周,等.荊芥的化學成分、藥理作用及臨床應用研究進展[J].中國藥房,2020,31(11):1397- 1402.
[ 3 ] 吳皓,李飛.中藥炮制學[M]. 2版.北京:人民衛生出版社,2016:225.
[ 4 ] 國家藥典委員會.中華人民共和國藥典:四部[S]. 2020年版.北京:中國醫藥科技出版社,2020:31-32,232.
[ 5 ] 高明亮,藍錦珊,單鳴秋,等.中藥炭藥研究進展與研究策略思考[J].南京中醫藥大學學報,2020,36(5):696-703.
[ 6 ] 黃瑤,張雪蘭,羅宇琴,等.山萸肉及其不同酒制品的UPLC特征圖譜建立及色度值的差異性研究[J].中國藥房,2021,32(2):206-213.
[ 7 ] 黃夢婷,潘玲,鄧李紅,等.基于UPLC指紋圖譜與色度值的桑白皮生品和炮制品的鑒別及炮制終點研究[J].中國藥房,2021,32(1):56-63.
[ 8 ] 楊麗,龔燚婷,許銘珊,等.基于“表里關聯”的大黃炭炮制過程顏色和成分變化關系研究[J].中草藥,2020,51(22):5705-5713.
[ 9 ] 孟慶安,劉恩順.實現中藥顏色客觀化表達的研究思路探討[J].天津中醫藥,2014,31(11):696-699.
[10] 張雪,李曉慶,王云,等.焦梔子炒制過程中HPLC圖譜變化與外觀顏色的動態關聯研究[J].中草藥,2018,49(17):4029-4037.
[11] 干麗,鐘如帆,魏梅,等.狗脊炮制工藝優選及色度值測定[J].中成藥,2020,42(9):2382-2388.
[12] 楊麗,溫雅心,劉洋,等.大黃炭加熱過程顏色特征與14種化學成分含量變化關系研究[J].中國中藥雜志,2020,45(17):4230-4237.
[13] 儲兆錚,王淑英,易劍平,等.荊芥炒炭成分變化趨勢初探[J].時珍國醫國藥,2019,30(8):1886-1888.
(收稿日期:2021-02-03 修回日期:2021-05-11)
(編輯:鄒麗娟)
