復合堿定向解聚綠竹堿木質素制備酚類化合物的研究
2021-07-21 01:34:40林秀華劉明華陳立暉呂源財
中國造紙學報 2021年2期
關鍵詞:催化劑
林秀華 劉明華,2,* 陳立暉 呂源財,2
(1.福州大學環境與資源學院,福建福州,350116;2.福建省生物質資源化技術開發基地,福建福州,350116)
近年來,隨著傳統化石能源危機以及環境污染等問題的日益嚴峻,人們逐漸把目光轉移到可再生能源上[1-2]。作為一類清潔且可再生的資源[3-4],生物質的資源化利用已成為當前研究熱點[2,5-6]。木質素是自然界中僅次于纖維素的第二大天然高分子材料,也是自然界中苯環含量最高的可再生資源,對其降解以生產芳香化學品無疑是未來木質素高值化利用的理想途徑[7-9]。然而,由于木質素存在構效關系不明晰、分子間高度集聚和化學性質非常穩定且不溶于大多數溶劑等特性,導致其在降解過程中面臨著降解效率低、產物不穩定以及工藝難控制等問題[10-11]。因此,開發木質素定向解聚的新工藝是解決木質素高值化應用瓶頸的重要方向[12-14]。
目前,木質素經過氧化、水熱、還原、裂解、酶解等反應可解聚為高附加值酚類化合物[15-19]。其中,水熱反應是一種很有前景的環境友好型熱處理技術,可斷裂木質素中的醚鍵和C—C鍵而保留其苯環結構,生成所需要的低分子酚類單體和二聚體。Gosselink等[20]研究了硬木木質素和麥秸木質素在超臨界二氧化碳/丙酮/水溶劑中的水熱降解反應,在溫度為300℃、壓力為10MPa時,獲得10%~12%產率的酚類化合物,但殘余焦炭高達51.5%;康世民[21]以堿木質素為原料,在300℃下水熱降解反應30min,木質素轉化率約50%。降解溫度過高往往會伴隨木質素的熱降解反應,酚類化合物會進一步分解生成氣體或者縮合形成焦炭[17,22],不利于木質素降解效率和酚類產物收率的提高。因此,需要尋找更加溫和的反應條件和催化劑體系,進一步提高酚類化合物的收率。相對于高溫(≥300℃)高壓(≥3MPa)等苛刻的反應條件,堿催化劑水熱降解法因具有經濟可行、操作簡便、高效等優勢而得到了廣泛關注。堿水熱降解不僅可有效破壞木質素中的醚鍵生成酚類化合物,還能抑制水熱反應中焦炭的形成[23]。
福建省竹資源豐富,為木質素的高值化利用提供了豐富的原料。綠竹是優良的筍竹兩用經濟竹種,福建省和廣東省是我國綠竹的中心產區。綠竹具有成林快、輪伐期短、產量高、筍質優、收益大等優勢,綠竹中纖維素、半纖維素和木質素的分離及其高值化利用是實現綠竹高效利用的關鍵。基于此,本課題組以綠竹堿木質素為原料,以低分子質量酚類化合物為目標產物,采用復合堿協同降解木質素,構建了一個高效、經濟、綜合的協同催化降解系統,在溫和的水熱條件下,通過調控復合堿的協同作用以實現C—O鍵和C—C鍵的選擇性斷裂,將木質素定向解聚為芳香結構小分子酚類化合物,并考察了復合堿對綠竹堿木質素降解效果的影響。
1 實驗
1.1 主要試劑
綠竹堿木質素由福建省三明市緣福生物質科技有限公司提供,粉碎過篩,干燥至質量恒定備用;HCl、H2SO4、NaOH、KOH、焦磷酸鈉、苯酚、MgO、Na2SO3、福林酚試劑等均為分析純;四氫呋喃(THF)和乙酸乙酯為色譜純。
1.2 主要儀器
紫外可見分光光度計,UV-1780,日本島津公司;凝膠滲透色譜儀(GPC),Waters2414,美國Waters公司;傅里葉變換紅外光譜儀(FT-IR),IR Prestige-21,美國Perkiin-Emler公司;全二維氣相色譜-高分辨飛行時間質譜聯用儀(GC-MS),Agilent 6890-59731,美國安捷倫科技有限公司。
1.3 木質素的提純
用20 wt%H2SO4溶液調節綠竹堿木質素使之pH值為2,靜置15~24 h后進行固液分離,分離的固相木質素離心水洗至中性。最后,將上述木質素進行冷凍干燥,密封備用。用元素分析儀(EA,Vario EL cube,德國Elementar公司)對綠竹堿木質素進行元素分析,得到C、H、O、N、S的質量百分比分別為54.25%、4.72%、35.52%、1.81%、0.84%,由此可得出,實驗用綠竹堿木質素的分子式為C9H9.93O4.42。根據GB/T 2677.2—2011、GB/T 742—2018、GB/T 2677.8—1994以及GB/T 10337—2008測定綠竹堿木質素的組成成分,結果如表1所示。

表1 綠竹堿木質素的組成成分Table 1 The component of Sinocalamus Oldhamialkali lignin
1.4 木質素降解
稱取2 g的木質素于50 mL燒杯中,加入與木質素質量比為1∶1的堿催化劑(復合堿中NaOH和堿性鹽質量比為3∶1)和20 g的蒸餾水配制成堿溶液,充分攪拌至木質素全部溶解。將溶液轉移至水熱反應釜,用氮氣吹掃趕出釜內空氣,于200℃條件下反應5 h。反應結束后,冷卻至常溫出料。實驗設置不添加催化劑的體系為空白組。
1.5 產物分離與處理
木質素降解產物先用1 mol/L HCl調節pH值至2后再離心分離。離心后的固體沉淀物冷凍干燥,稱量即得木質素殘渣的質量。液相部分用乙酸乙酯萃取3次,有機相除去溶劑后得到富含酚類化合物的產物。堿降解木質素流程及產物分離示意圖如圖1所示。
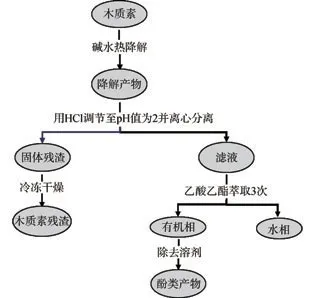
圖1 堿降解木質素流程及產物分離示意圖Fig.1 Schematic illustration of the alkali degradation procedure and product separation of lignin
1.6 分析方法
1.6.1 木質素及其降解產物酚羥基含量測定
木質素及其降解產物的酚羥基含量采用Folin-Ciocalteu法(福林酚法,簡稱FC法)測定[24-26]。以苯酚作為測定酚羥基含量的標準物質,根據苯酚在760 nm處的吸光度(A)與濃度(c,μmol/L)的關系做回歸方程:A=0.0101c+0.0122(相關系數R2=0.998),用該方法可測得綠竹堿木質素及其降解產物的酚羥基含量。
1.6.2 木質素的FT-IR分析
木質素的FT-IR譜圖表征采用傅里葉變換紅外光譜儀,掃描范圍400~4000 cm-l,掃描次數32次。
1.6.3 木質素的分子質量及其分布測定
先對木質素進行乙酰化預處理[27],將乙酰化木質素按5 mg/mL溶解在THF中。采用GPC測定乙酰化木質素的相對分子質量。GPC分析條件:聚苯乙烯(PS)為標樣,THF為流動相,流速1.0 mL/min,進樣量20μL。
1.6.4 木質素降解產物GC-MS分析
酚類產物的組成采用全GC-MS進行檢測。氣相檢測條件:色譜柱Rxi-5ms(30 m×0.25 mm×0.25μm),載氣為He,流速1.4 mL/min;初始柱溫50℃,保持1 min后,以10℃/min的速率升至300℃,保持10 min;進樣溫度280℃,進樣量1μL,分流比30∶1。質譜檢測條件:EI離子源,溫度250℃,電子能量70 eV。
1.7 實驗參數計算
木質素的降解率按式(1)計算[16,28],酚類化合物相對含量根據式(2)計算:
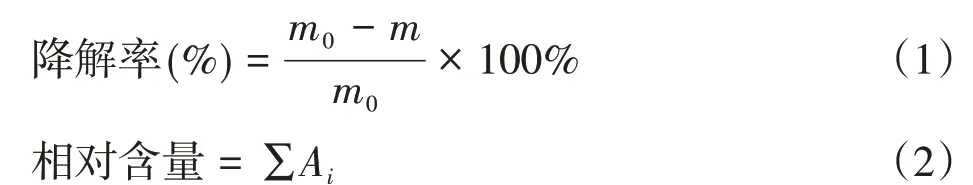
式中,m0、m分別表示降解前木質素質量(g)、降解過濾后的木質素殘渣質量(g);Ai指GC-MS檢測出的每種酚類所對應的相對峰面積。
2 結果與討論
2.1 堿催化劑對木質素降解率及產物酚羥基含量的影響
在200℃水熱條件下,分別采用MgO、NaOH和KOH等7種堿催化劑對木質素進行選擇性解聚,結果如圖2所示。從圖2可以看出,與空白組相比,添加不同的堿催化劑后,木質素降解率和產物酚羥基含量均有不同程度的提高。單堿催化劑中,NaOH催化降解木質素的效果較好,產物酚羥基含量約(1.8±0.1)mmol/g,相對于空白組提高了約76%,木質素降解率由(38.1±2.2)%提高到(79.1±1.3)%。與單堿催化劑相比,復合堿催化劑對木質素的降解效果更好,其中NaOH和Na2SO3組成的復合堿催化效果最佳,產物酚羥基含量為(3.5±0.07)mmol/g,相比空白組提高約244%,木質素降解率高達(83.1±1.2)%。上述結果表明,復合堿可進一步提高木質素的降解率并顯著增加產物酚羥基含量。這是由于NaOH和Na2SO3的協同作用更有助于木質素的解聚:一方面,復合堿中NaOH堿性較高,具有較強的堿性位點,從而促進木質素中醚鍵的斷裂,且木質素在NaOH溶液中的溶解性好,反應基本處于均相體系,降解更徹底,木質素殘渣率也較低[29]。此外,β-O-4醚鍵是木質素水熱降解過程中最容易被裂解的連接鍵,其斷裂的同時會生成酚鈉衍生物和瞬間被OH-中和的碳正離子,Na+通過與原木質素中的酚羥基發生化學反應,形成陽離子化合物而極化醚鍵,從而促進醚鍵的斷裂,釋放出酚羥基。因此,NaOH在復合堿降解木質素中具有較好的催化作用,表明堿的性質和強度會影響木質素的解聚效果。另一方面,在堿性條件下,Na2SO3和焦磷酸鈉的親核性遠大于NaOH。SO32-比OH-具有更強的親核作用,因而更易攻擊木質素苯環上的甲氧基,甲氧基的sp3雜化軌道電子云偏向氧原子,使甲氧基中的C—H鍵能減弱,從而使甲基脫落,酚羥基含量進一步增加[30]。因此,復合堿降解木質素后能生成更多的酚羥基。此外,在親核試劑SO32-的攻擊下,木質素大分子結構單元之間的α-烷基芳基醚鍵和β-O-4型連接鍵很容易發生斷裂,使木質素發生碎片化[31]。因此,在復合堿中NaOH和Na2SO3的協同作用下,木質素結構中的C—O鍵(以β-O-4為主)更易有效裂解和發生脫甲基反應,生成新的酚羥基結構,木質素降解率進一步提高,生成大量小分子的酚類產物。
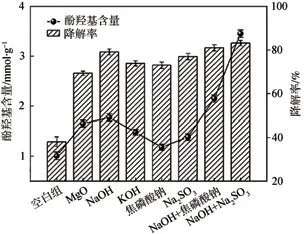
圖2 不同堿催化劑對木質素降解率及產物酚羥基含量的影響Fig.2 The effect of different alkali catalysts on the degradation efficiency of lignin and the phenolic hydroxyl content of the product
2.2 堿催化劑對木質素相對分子質量的影響
采用GPC對木質素及降解后溶于THF的殘余木質素相對分子質量進行測定。GPC測定結果表明,不同堿催化劑水熱降解木質素反應后,木質素的相對分子質量均有不同程度降低(見表2)。由表2可以看出,綠竹堿木質素的質均相對分子質量(Mw)在復合堿作用下,分別由11341降到1308和939,遠低于原料木質素的相對分子質量。通常認為,木質素中的芳基醚鍵含量越少,則相對分子質量越小;縮合結構越少,相對分子質量也越小[32]。這說明在復合堿催化降解過程中,木質素單元之間的連接鍵斷裂,木質素大分子變小,降解后生成大量小分子物質,這與酚羥基含量增加的結果一致。而空白組中降解產物的相對分子質量變化不大,說明產物仍然以大分子化合物為主。此外,原料木質素和殘余木質素的多分散系數也均小于2.0,說明該綠竹堿木質素和殘余木質素的相對分子質量分布區間較窄,趨于均一化。在堿性水熱降解作用下,木質素有效地斷裂結構中的C—O鍵,使其大分子碎片化、相對分子質量降低、相對分子質量的均一性提高。此外,實驗用綠竹堿木質素原料來自于一年生嫩綠竹,其相對分子質量和分子結構相對于其他種類木質素有較大的不同,更易被降解,也有利于降解后殘余木質素相對分子質量的均一化。

表2 綠竹堿木質素及不同堿催化劑降解后殘余木質素GPC分析Table 2 GPC analysis of the alkali lignin from Sinocalamus Oldhami and residual lignin from different alkali catalysts
2.3 復合堿降解木質素對降解產物分布的影響
2.3.1 FT-IR分析
綠竹堿木質素、單堿催化劑及復合堿催化劑降解后木質素殘渣的FT-IR譜圖如圖3所示。根據文獻[33-35]可分析確定FT-IR光譜中吸收峰的歸屬。3200~3650 cm-1為酚羥基的伸縮振動峰。由圖3可知,復合堿降解后木質素殘渣在該處的伸縮振動峰較寬,說明木質素的醚鍵斷裂,酚羥基的數量明顯增多。復合堿降解后木質素殘渣在2848~2950 cm-1處甲基、亞甲基的C—H伸縮振動峰明顯減弱,說明在降解過程中木質素苯環側鏈發生甲基、亞甲基脫離。由于木質素結構單元中側鏈C3結構中存在較多的甲基和亞甲基,C3側鏈的脫離意味著β-O-4醚鍵斷裂,可能形成了單酚類的化合物[36]。1700~1750 cm-1處的峰歸屬于C=O的特征伸縮振動峰,該峰只在綠竹堿木質素中出現;1215和1115 cm-1處的吸收峰是芳香醚C—O伸縮振動峰[33],木質素降解后該振動峰明顯減弱,這是降解過程中醚鍵斷裂導致,說明木質素成功被降解;木質素殘渣的酚羥基伸縮振動峰增大表明,在復合堿降解過程中,木質素分子中的β-O-4等結構發生斷裂生成了酚羥基,同時木質素中的甲氧基轉變為酚羥基[37]。1500~1600 cm-1處的吸收峰對應于芳環骨架伸縮振動峰,該峰在木質素降解前后沒有太大變化,說明在堿降解過程中,木質素的芳環結構沒有被破壞;750~860 cm-1處的吸收峰對應于芳香環C—H振動峰,堿催化降解木質素后,該振動峰響應強度增大,說明反應主要發生在苯環側鏈和其反應官能團上[38]。
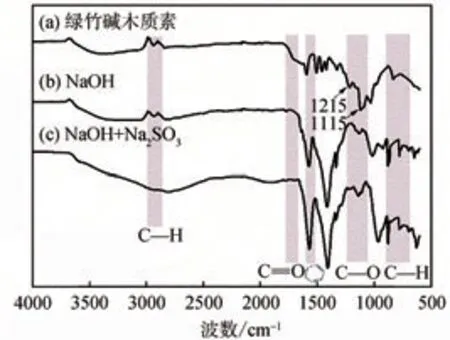
圖3 綠竹堿木質素、單堿催化劑及復合堿催化劑降解后木質素殘渣的FT-IR譜圖Fig.3 FT-IR spectra of alkali lignin from Sinocalamus Oldhami and residual lignin from single and composite alkali catalysts
2.3.2 GC-MS分析
木質素降解后主要酚類產物的GC-MS圖譜及分析結果見圖4和表3。由圖4和表3可知,在不使用催化劑水熱降解綠竹堿木質素的體系(空白組)中,木質素大分子很難被降解,因此酚類產物的相對含量較低且種類較少,相對含量僅為41.92%;在單獨NaOH降解體系中,木質素降解產物種類顯著增加,并含有大量的酚類物質,且酚類產物的相對含量(73.97%)明顯高于空白組體系,其中H型、G型和S型酚類物質的相對含量分別為29.62%、25.58%、17.00%,說明NaOH可以有效降解木質素。這是由于NaOH具有較強的堿水解作用,使木質素結構中的醚鍵斷裂生成酚類化合物;在單獨使用Na2SO3降解木質素的體系中,共產生6種酚類產物,酚類產物的相對含量為64.60%,其中H型、G型和S型酚類物質的相對含量分別為47.23%、14.03%、3.34%。相對于NaOH體系,單獨Na2SO3體系對產物的選擇性更好,可有效提高H型酚類產物的相對含量,同時G型和S型酚類產物的相對含量顯著下降,說明Na2SO3對于鍵能較大的鄰位甲氧基具有一定的脫除作用,有利于H型酚類物質的生成,但酚類產物總相對含量不高,這是由于Na2SO3難以有效破壞木質素大分子結構單元之間的醚鍵;在復合堿降解綠竹堿木質素體系中,主要產物是小分子酚類化合物,包括酚類物質單體和二聚體,酚類產物的相對含量高達86.85%,酚類產物共有10種,H型酚類物質相對含量最高(44.44%),G型酚類物質次之(23.85%),S型酚類物質最少(僅15.31%)。相比單獨的NaOH體系,NaOH+Na2SO3復合堿體系的酚類產物相對含量增加了12.88個百分點,并提高了H型酚類物質的相對含量,G型和S型酚類物質的相對含量降低。說明NaOH+Na2SO3復合堿可以高效降解木質素,并對綠竹堿木質素水熱降解生成酚類產物具有顯著的促進作用。這是由于NaOH和Na2SO3組成的復合堿具有強烈的堿活化催化作用和親核作用。在NaOH+Na2SO3復合體系中,Na+能夠極化木質素結構中的C—O鍵并降低其化學鍵的活化能,與此同時,體系中的OH-可奪取木質素分子酚羥基中的氫,引發一系列電子轉移過程[23];此時,NaOH+Na2SO3復合體系中的親核試劑SO32-可有效地攻擊鍵能減弱的醚鍵,導致木質素結構中醚鍵發生斷裂,進而定向解聚生成酚類產物[31];此外,Na2SO3的親核作用也能促使木質素更容易發生脫甲基作用,從而使木質素中甲氧基的甲基脫去而轉變成酚羥基[39],進一步促進酚類化合物的形成。FT-IR分析結果也表明,NaOH+Na2SO3復合堿體系中,木質素結構中醚鍵的伸縮振動峰明顯減弱。因此,NaOH和Na2SO3在木質素催化降解過程中具有良好的協同作用。苯酚(RT(保留時間,下同)=356 s)、4-乙基-苯酚(RT=520 s)和4-羥基-苯乙酮(RT=745 s)等對羥基苯酚衍生物的大量形成,說明經NaOH+Na2SO3復合堿水熱降解后,木質素結構中的C—O鍵被破壞,從而生成了大量小分子酚類物質[40]。單酚類化合物在總體酚類化合物的占比為96.26%,這是由于α-O-4、β-O-4和5-O-4醚鍵是木質素3個苯基丙烷結構單元之間主要的連接鍵,經過堿水熱反應后,醚鍵斷裂而產生芳基羥基、醇羥基和烷基等中間體,這些中間體進一步被破壞生成低分子質量的產物,包括酚類單體,如苯酚、4-乙基-苯酚和2-甲氧基-苯酚等[41]。NaOH+Na2SO3復合堿的加入促進了苯酚的生成,苯酚相對含量占總體酚類化合物的40.97%,證明NaOH+Na2SO3復合堿對苯環側鏈上甲氧基具有一定的脫除作用,還能促進木質素水熱降解生成的愈創木酚等含甲氧基支鏈的單酚類化合物轉化為苯酚等更為簡單的酚類化合物。
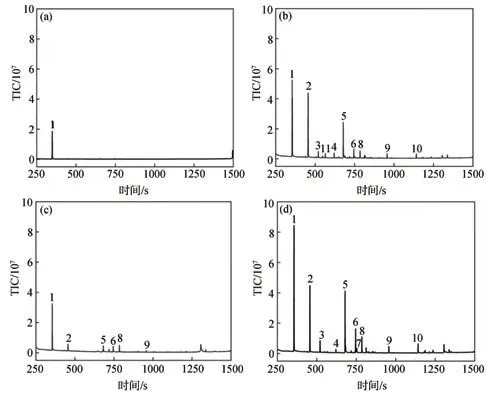
圖4 木質素降解后酚類產物的GC-MS譜圖Fig.4 GC-MS spectra of the phenolic compound from lignin degradation
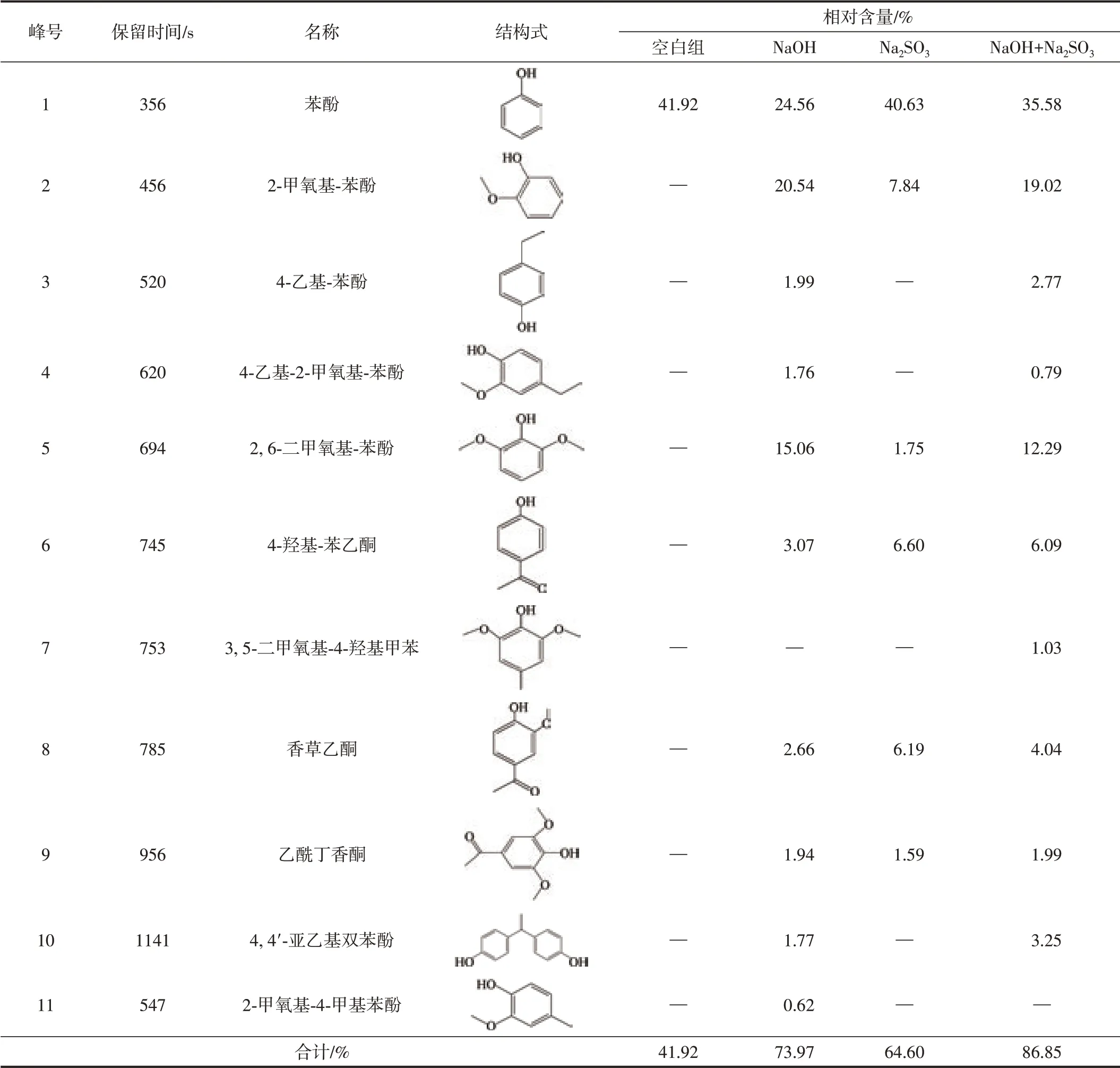
表3 木質素降解后主要酚類產物的GC-MS分析結果Table 3 GC-MS analysis results of main phenolic products after lignin degradation
3 結論
以綠竹堿木質素為原料,探究了不同堿催化劑降解木質素對酚類產物的影響。研究發現,在相對溫和的水熱體系中,采用復合堿催化劑可對綠竹堿木質素進行高效降解。NaOH和親核試劑Na2SO3組成的復合堿在降解綠竹堿木質素過程中具有協同作用,可有效地斷裂木質素結構中C—O鍵,使木質素發生定向解聚,降解率可達(83.1±1.2)%,并生成一系列低分子質量(<1000)的小分子物質,酚類產物的相對含量高達86.85%,實現了對酚類產物的高效選擇性。該木質素解聚工藝條件溫和,經濟可行,且降解效果理想,而該水熱體系能否適用于其他種類木質素的降解還有待進一步的探索。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
