不同液相色譜-串聯(lián)質(zhì)譜法檢測標(biāo)準(zhǔn)測量不確定度的評定
2021-07-22 00:43:24韓帥兵張耀中李向陽吳亞玉
農(nóng)藥科學(xué)與管理 2021年6期
韓帥兵,張耀中,于 淼,薛 雯,李向陽,吳亞玉,周 力
(山東省農(nóng)藥檢定所, 山東 濟(jì)南 250100)
隨著廣大人民群眾及政府部門對農(nóng)產(chǎn)品質(zhì)量安全重視程度的不斷加強(qiáng),各檢測機(jī)構(gòu)承接的農(nóng)產(chǎn)品中尤其是蔬菜水果中農(nóng)藥殘留檢測任務(wù)不斷增加,如何提供更加科學(xué)、準(zhǔn)確的檢測數(shù)據(jù)也成為各實(shí)驗(yàn)室關(guān)注的焦點(diǎn)。測量不確定度代表著檢測值的可信區(qū)間,是衡量實(shí)驗(yàn)室檢測質(zhì)量的重要指標(biāo),尤其是檢測結(jié)果在限量臨界值區(qū)間時(shí),不確定度更是合格性判定的重要參考依據(jù)[1-3]。因此,不確定度的評定逐漸受到各個(gè)實(shí)驗(yàn)室的重視。隨著不確定度評定相關(guān)標(biāo)準(zhǔn)及指南的更新或發(fā)布[4-5],不確定度的評定更加有據(jù)可循,測量不確定度的評定工作也相應(yīng)成為各實(shí)驗(yàn)室檢測工作的重要一環(huán)。當(dāng)前,液相色譜-串聯(lián)質(zhì)譜法作為一種應(yīng)用越來越普遍的農(nóng)藥殘留檢測方法,關(guān)于它的測量不確定度評定的研究雖有所報(bào)道[6-8],但這些報(bào)道大多不是建立在標(biāo)準(zhǔn)規(guī)定方法之上,對檢測實(shí)驗(yàn)室具體工作參考意義有限。
2021年3月,新的國家標(biāo)準(zhǔn)GB 23200.121-2021《食品安全國家標(biāo)準(zhǔn) 植物源性食品中331種農(nóng)藥及其代謝物殘留量的測定 液相色譜-質(zhì)譜聯(lián)用法》發(fā)布[9],此方法是建立在QuEChERS方法基礎(chǔ)上的多殘留檢測方法,與原國家標(biāo)準(zhǔn)GB/T 20769-2008《水果和蔬菜中450種農(nóng)藥及相關(guān)化學(xué)品殘留量的測定 液相色譜-串聯(lián)質(zhì)譜法》[10]相比,該方法用料量少、操作簡便,且涉及了很多原標(biāo)準(zhǔn)未涉及的農(nóng)藥代謝物,一定程度上來說,可以替代原國標(biāo)。這兩種檢測方法在前處理步驟上存在明顯差異,那么,在測量不確定度的評定過程及結(jié)果上是否也存在明顯差異,目前還沒有相關(guān)文獻(xiàn)報(bào)道。
本研究以檢測大白菜中吡蟲啉殘留量為例,按照標(biāo)準(zhǔn)規(guī)定的具體操作步驟及計(jì)算方法進(jìn)行檢測,依據(jù)不確定度評定相關(guān)標(biāo)準(zhǔn)及指南[4-5],分別評定了在相同檢測條件下,兩種檢測方法測量結(jié)果的不確定度,比較了二者在不確定度組成、最終結(jié)果、貢獻(xiàn)度分布及關(guān)鍵因素上的區(qū)別,并分析了產(chǎn)生原因,為標(biāo)準(zhǔn)方法的比較提供了數(shù)據(jù)依據(jù),同時(shí)也為農(nóng)藥殘留檢測實(shí)驗(yàn)室提供了不確定評定的參考范例。
1 實(shí)驗(yàn)部分
1.1 主要儀器與材料 超高效液相色譜-串聯(lián)質(zhì)譜儀(ESI);氮吹儀(N-EVAP112);旋轉(zhuǎn)蒸發(fā)儀(RV10 Control);移液器;石墨炭黑氨基SPE小柱(Carbon/NH2SPE,500mg,6mL);0.22μm有機(jī)濾膜、量筒、容量瓶等。
1.2 主要試劑 乙腈,色譜純;甲醇,色譜純;甲苯,分析純;氯化鈉,分析純;無水硫酸鎂,分析純;檸檬酸鈉,分析純;檸檬酸氫二鈉,分析純。
1.3 樣品前處理 樣品分別按照兩種標(biāo)準(zhǔn)規(guī)定的方法進(jìn)行前處理。
1.3.1 GB/T 20769-2008前處理 提取:準(zhǔn)確稱取20g(精確至0.01g)樣品于80mL離心管中,加入40mL乙腈,15 000r/min下勻漿1min,加入5g NaCl再勻漿1mL,離心后取上清液10mL,40℃氮吹至1mL后待凈化。
凈化:用5mL的V(乙腈) : V(甲苯)= 3:1混合液預(yù)淋Carbon/NH2柱后,加入待凈化液,用2mL上述混合液分3次洗滌離心管,將洗滌液轉(zhuǎn)入柱中,再用25mL淋洗液洗脫,收集所有洗脫液,旋轉(zhuǎn)濃縮近干,用5mL乙腈定容,過0.22μm有機(jī)濾膜后待測。
1.3.2 GB 23200.121-2021前處理 提取:準(zhǔn)確稱取10g(精確至0.01g)樣品于50mL離心管中,加入10mL乙腈、4g無水硫酸鎂、1g氯化鈉、1g檸檬酸鈉、0.5g檸檬酸氫二鈉及1顆陶瓷均質(zhì)子,蓋上離心管蓋,劇烈震蕩1min 后4 200r/min離心5min。
凈化:吸取6mL上清液至內(nèi)含900mg無水硫酸鎂、150mgPSA的離心管中,渦旋混勻1min。4 200r/min離心5min,吸取上清液過0.22μm有機(jī)濾膜后待測。
1.4 樣品檢測 前處理后的樣品經(jīng)UPLC-MS/MS檢測,兩種檢測標(biāo)準(zhǔn)采取相同的儀器方法,參考儀器條件如下:
色譜柱:Shim-pack XR-ODS Ⅲ(2.0mm I.D.×50mm,1.6μm)
流動(dòng)相:A:甲醇,B:5 mM醋酸銨+0.02%甲酸水溶液;柱溫:40℃;進(jìn)樣量:0.5μL
流速及梯度:(表1)
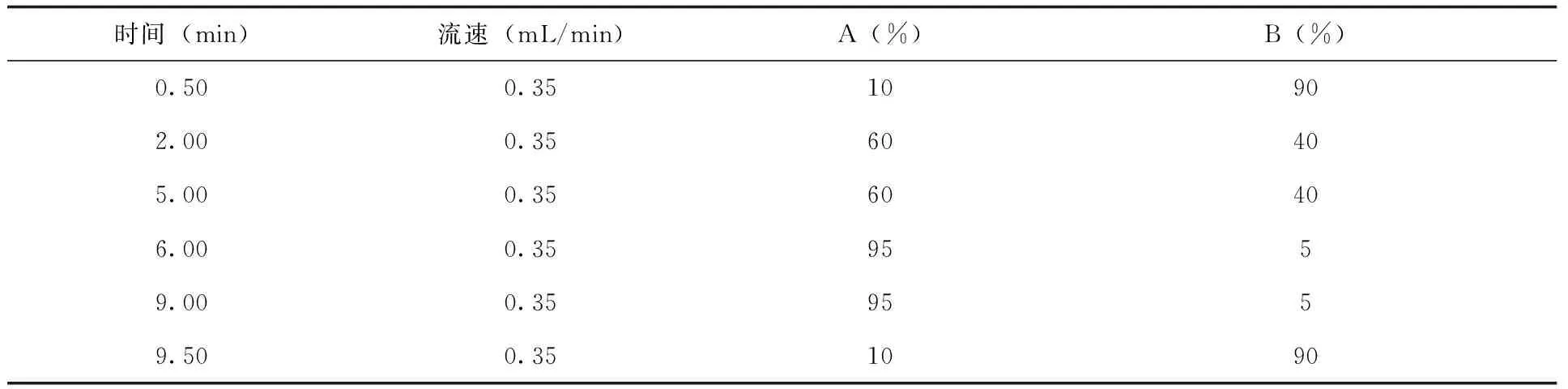
表1 流動(dòng)相流速及梯度設(shè)置
質(zhì)譜儀條件:離子源:ESI源,正離子模式;檢測方式:MRM;電離電壓:4.5kV;碰撞氣及壓力:Ar,0.23MPa;霧化氣流速:3.0L/min;檢測器電壓:1.74kV;干燥氣流速:10.0mL/min;加熱氣流速:10.0mL/min;定量離子:256.10-209.10,碰撞電壓-14V;定性離子:256.10-175.10,碰撞電壓-16V。
1.5 數(shù)學(xué)模型的建立 根據(jù)兩種檢測標(biāo)準(zhǔn)規(guī)定的方法,兩種方法的殘留量(mg/kg)計(jì)算分別見公式(1、2)。
(1)
(2)
其中,公式(1)為GB/T 20769-2008計(jì)算公式,公式(2)為GB 23200.121-2021計(jì)算公式。式中,ω為樣品中被測農(nóng)藥組分的含量(mg/kg);Ai為樣品中被測農(nóng)藥組分的峰面積;As為標(biāo)準(zhǔn)溶液中被測農(nóng)藥組分的峰面積;V1為提取溶液總體積(mL);V2為吸取出用于檢測的提取溶液體積(mL);V3為樣品定容體積(mL);m為樣品取樣量(g);ρ為標(biāo)準(zhǔn)溶液中農(nóng)藥的含量(mg/L)。
2 結(jié)果與討論
2.1 不確定度來源分析 通過對檢測過程的分析,兩個(gè)標(biāo)準(zhǔn)的檢測都有4個(gè)主要步驟:標(biāo)準(zhǔn)溶液配制、稱樣、前處理和上機(jī)檢測,據(jù)此可將引入不確定度的來源細(xì)化為:所購買標(biāo)準(zhǔn)溶液、標(biāo)準(zhǔn)溶液配制、稱樣、前處理過程、儀器穩(wěn)定性。另外,在實(shí)際檢測工作中,還需要考慮方法精密度對測定結(jié)果的系統(tǒng)性影響,由此可以得到不確定度來源分析魚骨圖(圖1)。

圖1 不確定度來源分析魚骨圖
其中,對于GB 23200.121-2021來說,樣品前處理過程中并不包含分取和定容的過程。綜合公式(1、2)及(圖1),可以判定檢測結(jié)果的不確定度主要來源包括:標(biāo)準(zhǔn)溶液質(zhì)量濃度(ρ),樣品進(jìn)樣峰面積(Ai),標(biāo)準(zhǔn)溶液峰面積(As),樣品的最終定容體積(V),樣品稱樣量(m)及方法精密度(frec),由此可根據(jù)公式(3)得到合成不確定度urel(ω):
即:urei(ω)=
(3)
2.2 相對不確定度的評定
2.2.1 標(biāo)準(zhǔn)溶液引入的不確定度 兩種檢測方法所用標(biāo)準(zhǔn)溶液均為用購買的吡蟲啉有證標(biāo)準(zhǔn)溶液稀釋后得到,因此,由標(biāo)準(zhǔn)溶液引入的不確定度是相同的,包括所購標(biāo)準(zhǔn)溶液urel(ρ1)和配制過程引入urel(ρ2),均為B類評定。
2.2.1.1 購買標(biāo)準(zhǔn)溶液引入的不確定度 本研究所購買吡蟲啉標(biāo)準(zhǔn)溶液質(zhì)量濃度為1 000mg/L,標(biāo)準(zhǔn)物質(zhì)證書顯示其擴(kuò)展不確定度為7mg/L(包含因子k=2),則所購標(biāo)準(zhǔn)溶液引入的相對不確定度為:urel(ρ1)=0.35%。
2.2.1.2 標(biāo)準(zhǔn)溶液配制引入的不確定度 本研究最終所用質(zhì)量濃度為0.2mg/L的標(biāo)準(zhǔn)工作溶液是使用質(zhì)量濃度為1 000mg/L的標(biāo)準(zhǔn)溶液用乙腈2次稀釋后配制而得:先用1mL A級分度吸量管移取0.25mL 1 000mg/L標(biāo)準(zhǔn)溶液,用乙腈定容到25mL,配制得1.0mg/L標(biāo)準(zhǔn)溶液;再用1mL A級分度吸量管移取1.0mL 1.0mg/L標(biāo)準(zhǔn)溶液,用空白樣品溶液定容到5mL,得到0.2mg/L的標(biāo)準(zhǔn)工作溶液。在標(biāo)準(zhǔn)溶液的配制過程中,引入不確定度的因素應(yīng)該包括溫度變化對試劑膨脹的影響,及配制過程中所使用玻璃儀器的允差及讀數(shù)誤差。
實(shí)驗(yàn)室溫度一般控制在(20±5)℃,乙腈的膨脹系數(shù)為0.001 37℃-1,服從矩形分布,由此,由溫度效應(yīng)引入的相對不確定度可由公式(4)計(jì)算而得:
(4)
配制過程中所用玻璃儀器引入的不確定度主要由允差和讀數(shù)重復(fù)性偏差組成,每個(gè)分量可用公式(5)計(jì)算:
(5)
式中aL為玻璃儀器的最大允許誤差或讀數(shù)重復(fù)性偏差,其中,1mL A級分度吸量管精度較高,讀數(shù)重復(fù)性引入不確定度可以忽略不計(jì),允許誤差為±0.008mL[11];25mL容量瓶允許誤差為±0.03mL,重復(fù)性偏差為±0.01mL;5mL容量瓶允許誤差為±0.02mL,重復(fù)性偏差為±0.01mL;服從三角形分布,k取 ;V為所用體積。由此,在標(biāo)準(zhǔn)溶液配置過程中每次使用玻璃儀器由誤差、偏差及溫度引入的不確定度(表2)。
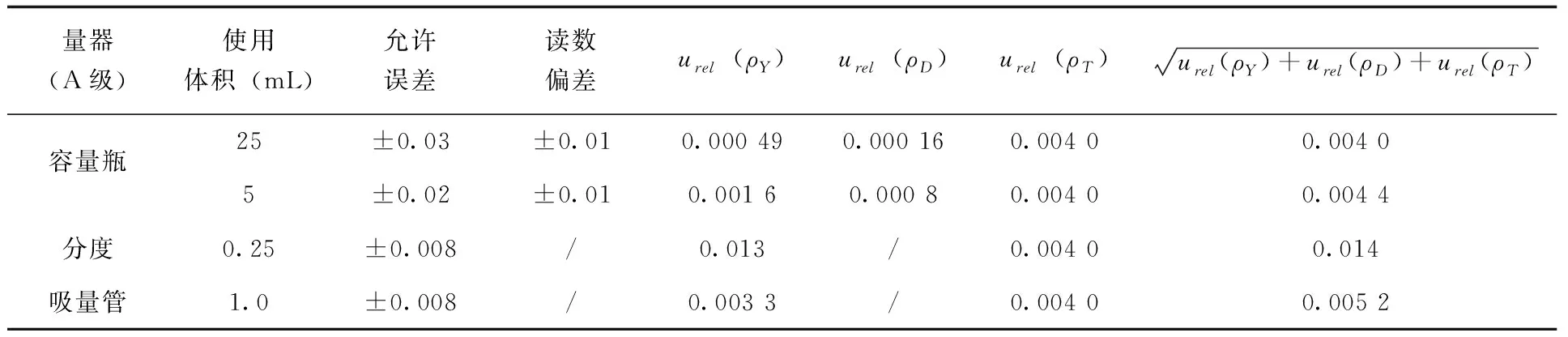
表2 標(biāo)準(zhǔn)溶液配制中玻璃儀器使用引入的不確定度
將上述不確定度合并,得到標(biāo)準(zhǔn)溶液配制過程引入的不確定度urel(ρ2)公式(6)。
(6)
據(jù)此,計(jì)算標(biāo)準(zhǔn)溶液所引入的不確定度urel(ρ)公式(7) 。
(7)
2.2.2 樣品稱量引入的不確定度 本研究中,兩種檢測方法稱樣時(shí)所用天平相同,均為精度為0.01g電子天平,樣品均勻性及稱量重復(fù)性引入不確定度均可忽略不計(jì)。該級別天平在量程內(nèi)允許的最大誤差為±0.01g,則由稱量誤差引入的不確定度服從矩形分布(B類評定),兩種方法稱樣量有所不同,則GB/T 20769-2008中由稱量引入不確定度urel(mⅠ)計(jì)算公式(8),GB 23200.121-2021中由稱量引入不確定度urel(mⅡ)計(jì)算公式(9)。
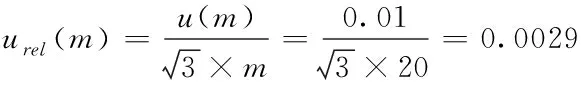
(8)
(9)
2.2.3 樣品前處理引入的不確定度 前處理所引入的不確定度主要來源于添加提取試劑、移取及凈化后定容時(shí)的玻璃器皿的允差及溫度變化引起試劑膨脹,均為B類評定。
2.2.3.1 GB/T 20769-2008前處理引入的不確定度 由1.3.1可知,對于GB/T 20769-2008來說,可以引入不確定度的步驟所用器皿包括:添加40mL乙腈所用50mL量筒,允許誤差為±0.50mL,讀數(shù)重復(fù)性偏差為±0.10mL;移取提取液所用10mL單標(biāo)線吸量管,A級允許誤差為±0.020mL,讀數(shù)重復(fù)性偏差可以忽略;最終定容所用5mL單標(biāo)線吸量管,A級允許誤差為±0.015mL,讀數(shù)重復(fù)性偏差可以忽略,均服從三角分布。應(yīng)用公式(4、5)可計(jì)算在前處理過程中每次使用玻璃儀器由誤差、偏差及溫度引入的不確定度(表3)。
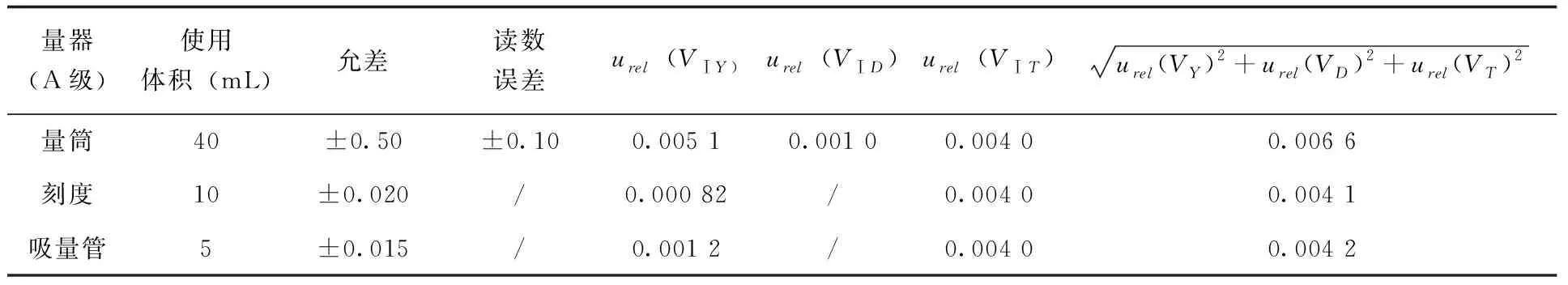
表3 GB/T 20769-2008前處理過程玻璃儀器使用引入不確定度
將上述不確定合并,得到前處理過程引入的不確定度urel(VⅠ)公式(10)。
(10)
2.2.3.2 GB 23200.121-2021前處理引入的不確定度 由1.3.2可知,對于GB 23200.121-2021來說,可以引入不確定度的步驟所用器皿只有在加入10mL乙腈時(shí)所用規(guī)格為10mL的量筒,其允許誤差為±0.10mL,讀數(shù)重復(fù)性偏差為±0.02mL;雖然在后續(xù)步驟中有提取液的取用,但這一步驟并不需精確量取,且不參與最終結(jié)果的計(jì)算,由此可以計(jì)算前處理過程引入的不確定度urel(VⅡ)公式(11)。
(11)
2.2.4 儀器檢測引入的不確定度 由2.1可知,儀器檢測引入的不確定度可以分為標(biāo)準(zhǔn)溶液峰面積引入的不確定度和樣品溶液峰面積引入的不確定度。由于超高效液相色譜-串聯(lián)質(zhì)譜儀靈敏度較高,可以認(rèn)為儀器穩(wěn)定性即測定值的分散性就是構(gòu)成測量峰面積的不確定度的主要來源,其他因素可以忽略不計(jì)。此類不確定度可以在重復(fù)性條件下對同一被測量對象獨(dú)立重復(fù)觀測多次,最后采用統(tǒng)計(jì)方法進(jìn)行計(jì)算,即進(jìn)行A類評定。在重復(fù)性條件下,對標(biāo)準(zhǔn)溶液和樣品進(jìn)行分別進(jìn)樣6次,得到6次進(jìn)樣的峰面積,根據(jù)貝塞爾公式(12)計(jì)算標(biāo)準(zhǔn)偏差,標(biāo)準(zhǔn)不確定度根據(jù)公式(13)計(jì)算。
(12)
(13)
根據(jù)方法(1、2)的標(biāo)樣溶液和樣品峰面積檢測結(jié)果,分別計(jì)算得到相應(yīng)的標(biāo)準(zhǔn)不確定度及相對標(biāo)準(zhǔn)不確定度(表4)。
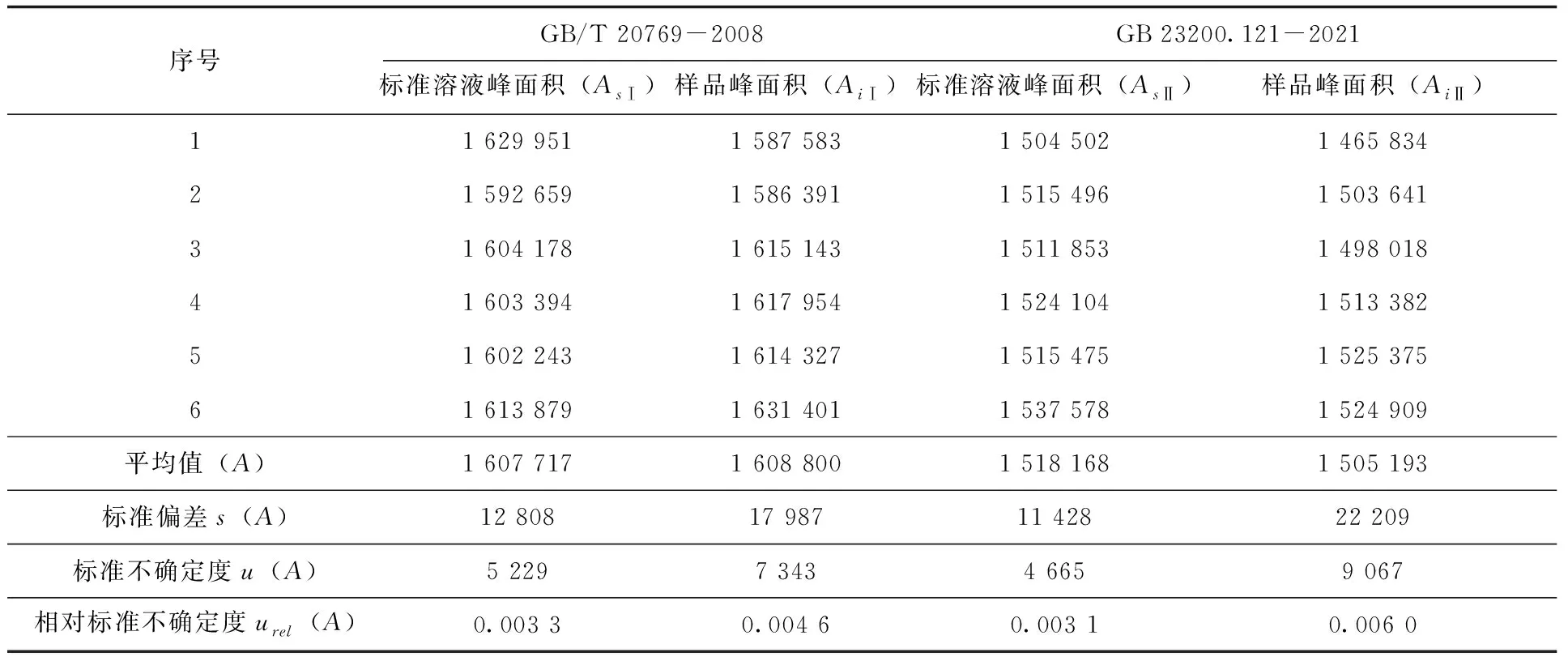
表4 標(biāo)準(zhǔn)溶液和樣品峰面積檢測結(jié)果及不確定度(n=6)
2.2.5 方法精密度引入的不確定度 由方法精密度引入的不確定度符合A類評定,本研究通過檢測多個(gè)空本樣品添加標(biāo)準(zhǔn)溶液進(jìn)行評定。按照GB/T 20769-2008及GB 23200.121-2021的稱樣量分別制備5個(gè)添加樣品并在重復(fù)性條件下進(jìn)行檢測,添加濃度均為0.2mg/kg,根據(jù)檢測結(jié)果應(yīng)用公式(12、13)計(jì)算相應(yīng)標(biāo)準(zhǔn)偏差及標(biāo)準(zhǔn)不確定度,最終計(jì)算得到相對標(biāo)準(zhǔn)不確定度(表5)。
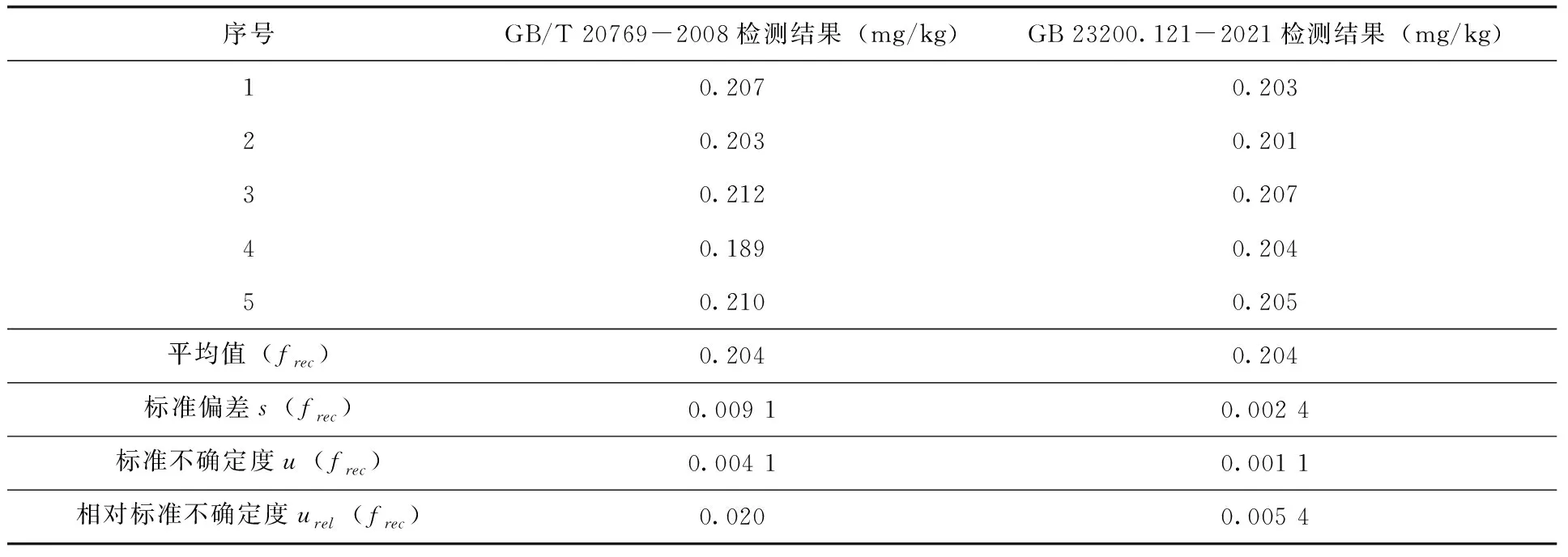
表5 添加回收樣品檢測結(jié)果及不確定度(n=5)
2.2.6 相對標(biāo)準(zhǔn)不確定度的合成 綜合上述得到的各不確定度分量,通過公式(3)可計(jì)算得到兩個(gè)方法的合成相對標(biāo)準(zhǔn)不確定度urel(ω),具體各分量不確定度及合成相對不確定度計(jì)算結(jié)果(表6)。
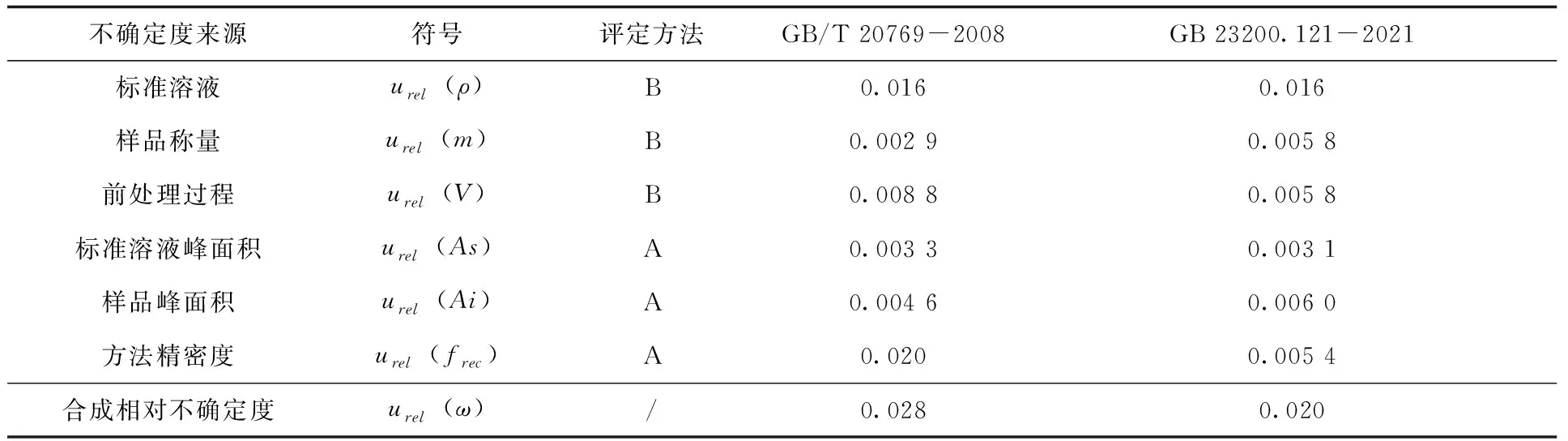
表6 各分量不確定度及合成相對不確定度
由結(jié)果可知,對于本次實(shí)驗(yàn)來說,兩種方法的合成相對標(biāo)準(zhǔn)不確定度均比較低,但依舊存在一定的差異,GB/T 20769-2008明顯高于GB 23200.121-2021,主要差別出現(xiàn)在方法精密度上,其他分量引入的不確定度相差并不多。分析原因應(yīng)該是由于GB/T 20769-2008的前處理步驟相對復(fù)雜,需要經(jīng)過濃縮—凈化—濃縮—定容等一系列操作,造成方法精密度較方法二更低,從而提高了不確定度。
對于每個(gè)分量對合成相對標(biāo)準(zhǔn)不確定度的貢獻(xiàn)度而言,GB/T 20769-2008結(jié)果中,貢獻(xiàn)度最高的是方法精密度(51.7%)和標(biāo)準(zhǔn)溶液(33.1%);其他分量的貢獻(xiàn)度都較低;GB 23200.121-2021結(jié)果中貢獻(xiàn)度最高的是標(biāo)準(zhǔn)溶液(64.3%),其他各分項(xiàng)貢獻(xiàn)度相對較低且比較平均。由此可見,對于兩種檢測方法來說,標(biāo)準(zhǔn)溶液的不確定度都是影響檢測結(jié)果不確定度的關(guān)鍵因素,而對于GB/T 20769-2008來說,精密度則是另一個(gè)關(guān)鍵因素。
雖然從本次實(shí)驗(yàn)的評定結(jié)果來看,在不確定度的表現(xiàn)上,GB 23200.121-2021確實(shí)優(yōu)于GB/T 20769-2008,但這一結(jié)果僅是反應(yīng)吡蟲啉一種農(nóng)藥在大白菜這一種基質(zhì)中的情況,并不能得到標(biāo)準(zhǔn)所涉及的所有農(nóng)藥在各種基質(zhì)中均符合這一規(guī)律的結(jié)論。這是因?yàn)椋瑑煞N檢測方法樣品的最終凈化效果是差別較大的,GB/T 20769-2008對樣品的凈化效果要明顯優(yōu)于GB 23200.121-2021,在這種情況下,對于復(fù)雜基質(zhì),比如含色素較多的綠色蔬菜,含其他雜質(zhì)較多的鱗莖類蔬菜等,GB 23200.121-2021是否還能保持優(yōu)于GB/T 20769-2008的精密度,進(jìn)樣溶液雜質(zhì)的增多是否會(huì)影響儀器檢測峰面積的重復(fù)性等,這些因素都會(huì)影響最終不確定度的評定結(jié)果,還需要通過進(jìn)一步的實(shí)驗(yàn)具體分析。
2.3 檢測結(jié)果擴(kuò)展不確定度的表示 根據(jù)測量不確定度評定指南對一般實(shí)驗(yàn)室的要求,在置信概率p=95%時(shí),取測量結(jié)果的擴(kuò)展不確定度包含因子k=2,由此得到,對于一個(gè)吡蟲啉在大白菜樣品中殘留量的檢測結(jié)果為MRL值[12](0.2mg/kg)附近的樣品來說,用標(biāo)準(zhǔn)方法GB/T 20769-2008和GB 23200.121-2021分別進(jìn)行檢測,其結(jié)果的相對擴(kuò)展不確定度分別為5.6%和4.0%。
3 結(jié)論
本研究評定了使用兩種液相色譜-串聯(lián)質(zhì)譜法檢測吡蟲啉在大白菜中殘留量時(shí)結(jié)果的不確定度。通過實(shí)驗(yàn)及計(jì)算可知,由于檢測方法更加簡便易行,方法重現(xiàn)性更好,新檢測標(biāo)準(zhǔn)GB 23200.121-2021的結(jié)果不確定度要小于原方法GB/T 20769-2008。但是,本研究只針對吡蟲啉一種農(nóng)藥在大白菜基質(zhì)中,對于更多種農(nóng)藥在其他基質(zhì)尤其是復(fù)雜基質(zhì)中的不確定度是否符合這一規(guī)律還有待更進(jìn)一步的研究。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
當(dāng)代陜西(2019年8期)2019-05-09 02:22:48
動(dòng)漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
意林原創(chuàng)版(2016年10期)2016-11-25 10:28:30
海峽科技與產(chǎn)業(yè)(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
專用汽車(2016年4期)2016-03-01 04:13:43
