水泥礦物體系誘導(dǎo)期的水化進(jìn)程及機(jī)理的研究進(jìn)展
2021-08-10 01:38:46胡匡藝儲靜遠(yuǎn)艾曉晶蓋琳琳盛嘉誠
硅酸鹽通報 2021年7期
官 敏,胡匡藝,于 濤,儲靜遠(yuǎn),艾曉晶,常 鄭,蓋琳琳,盛嘉誠
(中國建材國際工程集團(tuán)有限公司,上海 200063)
0 引 言
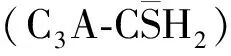
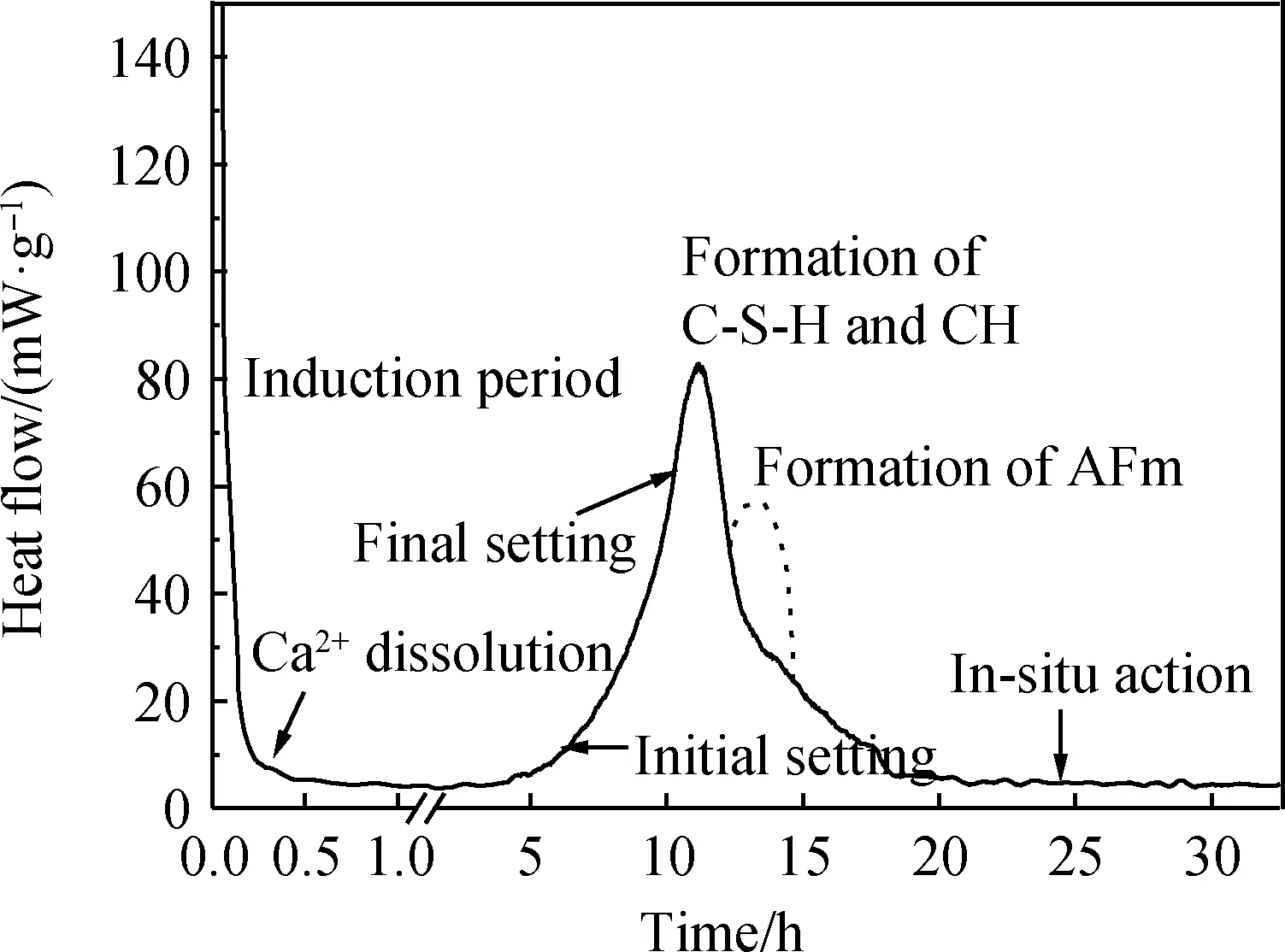
圖1 水泥水化進(jìn)程的劃分[3]Fig.1 Hydration process division of Portland cement[3]
1 水化作用的熱力學(xué)原理
水泥水化作用的基礎(chǔ)是溶解-沉淀過程,未水化相與水化產(chǎn)物之間如果沒有溶液中的離子擴(kuò)散是不能發(fā)生轉(zhuǎn)化的。水化進(jìn)程中的水化產(chǎn)物必須具有比未水化相更低的溶解度。在氧化鈣-氧化鋁-水(CaO-Al2O3-H2O)三元體系中,溶解度最低的是3CaO·Al2O3·6H2O(C3AH6)。但是C3AH6的形成慢于溶解度更高的2CaO·Al2O3·8H2O(C2AH8) 以及4CaO·Al2O3·13H2O(C4AH13),因此后者首先生成,提高溫度則可以促進(jìn)向C3AH6的轉(zhuǎn)變[4],這表明結(jié)合熱力學(xué)與動力學(xué)或許可以預(yù)測產(chǎn)物的生成。
但不同條件下物相溶解度的差異對研究多相系統(tǒng)又是不足的,例如對硅酸三鈣-硅酸二鈣(C3S-C2S)二元體系的研究[5]。C3S與C2S的溶解度均高于水化硅酸鈣(C-S-H),但是在均勻混合的C3S-C2S二元體系中,只要C3S正在水化,C2S便無法溶解。這是因?yàn)镃3S水化時溶液中的離子濃度高于C2S的溶解度。所以,了解物相的水化速率是必要的。
水化作用是一個復(fù)雜過程,至少涉及到兩種固相(一種未水化相(反應(yīng)相)和一種水化產(chǎn)物相(生成相))和一種液相。未水化相的溶解存在溶解速率Rd,水化產(chǎn)物的沉淀存在沉淀速率Rp。水化是一種界面反應(yīng),因此水化速率還與固相與液相的接觸面積S有關(guān)。從熱力學(xué)角度出發(fā),偏離平衡越遠(yuǎn),吉布斯自由能的絕對值ΔG越大,Rd和Rp越高。此外,溶液中的一些離子可能對表面反應(yīng)具有特定的影響。在各向同性材料的情況下,t時刻的溶解或沉淀速率可寫為[6]:
R(t)=rif(ΔG(t),[ions], …)S(t)
(1)
式中:R(t)為t時刻的溶解或沉淀速率,mol·s-1;rif為溶解或沉淀的界面速率,mol·m-2·s-1;[ions]為任意離子濃度,mol·L-1;S(t)為溶解或生成的表面積,m2。
rif與S均隨時間變化。考慮到溶解時固體表面積的減少以及沉淀時固體表面積的增加,在水化進(jìn)程中溶解界面速率和沉淀界面速率也是不同的,但在大部分時間里,溶解速率近似等于沉淀速率,即:
Rp?Rd
(2)
由于在水化開始時溶解的表面積Sp為0,而在水化結(jié)束時生成的表面積Sd為0,為了滿足式(2),反應(yīng)相溶解的界面速率與生成相沉淀的界面速率的比值(rifd/rifp)必須持續(xù)增大。水化開始時,溶液中離子濃度近似于反應(yīng)相的溶解度,而在水化結(jié)束前,溶液中的離子濃度接近生成相的溶解度。該過程被Barret[5]命名為水化作用的動力學(xué)過程。
水化作用的開始階段很特別,除晶核的形成外,生成相還不存在,此時僅存在反應(yīng)相單獨(dú)溶解使足夠的離子進(jìn)入液相誘導(dǎo)新相(生成相)的產(chǎn)生。實(shí)際上,如果生成溶解度較低的新相使體系的自由能降低,那么在水化產(chǎn)物與低自由能的溶液之間會形成新的界面,形成界面所需的自由能來自溶液對于生成相的過飽和。根據(jù)經(jīng)典形核理論,形成可以生長的穩(wěn)定晶核所需的誘導(dǎo)時間tip取決于溶液過飽和度β(離子活度積Iap與平衡溶度積Ksp的比值) 以及晶體與溶液間的界面能γ[6],如式(3)所示:
(3)
式中:f為形核系數(shù);Ω為摩爾體積;κ為玻爾茲曼常數(shù);T為形核溫度;K0為動力學(xué)常數(shù)。
溶液過飽和度越高,形核所需的誘導(dǎo)時間越短。如果反應(yīng)相的溶解速率較高,并且反應(yīng)相的溶解度高于生成相的溶解度,則可以更快達(dá)到生成相所需的過飽和度。C-S-H在C3S水化進(jìn)程中形核的時間極短,幾乎無法測量[7]。相反,C2S在氫氧化鈣(CH)溶液中發(fā)生水化作用時,誘導(dǎo)期長達(dá)數(shù)十分鐘,因此可以觀察C-S-H的形核過程[8-9]。在鋁酸一鈣(CA)水化進(jìn)程中,CaO·Al2O3·10H2O(CAH10)形核所需的時間可能更長[10-11]。

這些理論使理解和預(yù)測愈加復(fù)雜的膠凝材料體系隨時間的水化進(jìn)程成為可能。綜上所述,大多數(shù)水泥水化進(jìn)程可以滿足下式[6]:
R(t)=rifd(t)Sd(t)=rifp(t)Sp(t)
(4)
式中:rifd(t)為t時刻未水化相溶解的界面速率;Sd(t)為t時刻未水化相溶解的表面積;rifp(t)為t時刻水化產(chǎn)物相沉淀的界面速率;Sp(t)為t時刻水化產(chǎn)物相生成的表面積。
2 C3S體系的溶解
作為水泥中占比最大的組分,C3S的水化受到了極大的關(guān)注。C3S體系在水化進(jìn)程中重要的特征之一是在加入水之后C3S反應(yīng)速率迅速降低,導(dǎo)致水化過程減緩,一段時間后反應(yīng)速率再次提升。誘導(dǎo)期具有重要的實(shí)際意義,可以提供在終凝之前開始運(yùn)輸和澆筑混凝土的時間。然而,誘導(dǎo)期在實(shí)際放熱曲線中通常表現(xiàn)為最小值,如圖2 C3S體系的水化放熱曲線所示,并非圖1中所示的明顯平坦階段。多年來,C3S水化的誘導(dǎo)期一直是學(xué)界關(guān)注的問題,并且許多學(xué)者提出了不同假說來解釋其成因。其中介穩(wěn)層假說和緩慢溶解假說受到廣泛認(rèn)同,這兩種假說在大部分研究中具有較強(qiáng)的合理性。
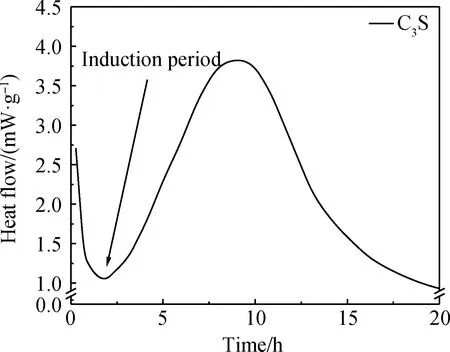
圖2 C3S體系的水化放熱曲線[11]Fig.2 Heat flow curve of C3S system hydration[11]
2.1 介穩(wěn)層假說(metastable barrier hypothesis)
Stein[13]及Jennings[14]等認(rèn)為,C3S體系早期水化速率降低是由于顆粒表面快速生成薄且連續(xù)的介穩(wěn)水化硅酸鈣層而引起的,這層水化產(chǎn)物被稱作介穩(wěn)態(tài)C-S-H (C-S-H(m))[15]。
C-S-H(m)通過阻礙未水化的C3S顆粒接觸水,或通過阻礙離子由C3S顆粒表面擴(kuò)散,從而抑制C3S體系的早期水化。在誘導(dǎo)期結(jié)束時,C-S-H(m)與溶液維持溶解平衡。
介穩(wěn)層假說表明C-S-H(m)將C3S顆粒與溶液完全隔離,而且與溶液達(dá)到溶解平衡。但是,關(guān)于誘導(dǎo)期如何結(jié)束的機(jī)理則并不明晰。誘導(dǎo)期結(jié)束的準(zhǔn)確時刻具有可重復(fù)性表明此時一定存在某種關(guān)鍵作用。換句話說,在誘導(dǎo)期結(jié)束時一定存在某種反應(yīng),使得C-S-H(m)最終變得不再穩(wěn)定[15-16]。事實(shí)上,由圖2可知,盡管誘導(dǎo)期內(nèi)C3S水化放熱速率大幅降低,但并不為零。核反應(yīng)分析(nuclear resonance reaction analysis, NRRA)結(jié)果可知[17],隨時間的推進(jìn)氫元素逐漸侵入固相內(nèi)部。氫元素侵入深度的增加表明,即使處于誘導(dǎo)期內(nèi),C3S的水化仍在進(jìn)行。而且隨著時間的推進(jìn),固相表面C-S-H的組成是變化的。

2.2 緩慢溶解假說(slow dissolution step hypothesis)
另一些學(xué)者認(rèn)為,雖然介穩(wěn)層假說可以解釋部分實(shí)驗(yàn)結(jié)果,但仍存在一些重要的問題未解決。一方面,現(xiàn)今的技術(shù)并不能直接觀察到C-S-H(m),如文獻(xiàn)[22]所述,水泥顆粒表面存在蝕坑,其鋒利的邊緣使C-S-H(m)不太可能存在。另一方面,目前普遍認(rèn)為最早生成的水化產(chǎn)物是C-S-H,但C-S-H的晶體結(jié)構(gòu)與C3S的晶體結(jié)構(gòu)完全不同,因此C-S-H在C3S表面很難致密附著,在顆粒表面多為孤立的團(tuán)塊存在[6],形貌如圖3所示。

圖3 水泥水化360 min后Alite顆粒表面上C-S-H的 SEM照片[6]Fig.3 SEM image of C-S-H on the surface of Alite grains after cement hydration for 360 min[6]
C3S水化進(jìn)程中所形成的初始水化產(chǎn)物并不足以抑制C3S水化的進(jìn)行。因此,Barret等[23-24]最早提出C3S表面與水接觸生成表面羥基化層(superficially hydroxylated layer),表面羥基化的C3S其表觀溶解度遠(yuǎn)小于C3S的理論溶解度,并且當(dāng)溶液中CH濃度增加時,C3S的溶解進(jìn)一步受到抑制。C3S溶出速率的結(jié)果表明[25],在沒有C-S-H的形核和生長等因素的干擾下,溶液中鈣元素和硅元素的濃度始終以3 ∶1的比例上升。因此,C3S體系水化誘導(dǎo)期的產(chǎn)生可以依據(jù)溶解-沉淀過程來解釋,一些學(xué)者[7,26-29]在此基礎(chǔ)上提出了緩慢溶解假說。
許多礦物相在水中的溶解速率與溶液的飽和狀態(tài)間沒有絕對的線性關(guān)系,換句話說,礦物相遇水后溶解速率下降是共價鍵礦物的常態(tài)[6,30-32]。新相的產(chǎn)生總是伴隨著過冷或過飽和現(xiàn)象,需要一定的過冷度或過飽和度來提供最初形核的自由能,而在一定溫度下自由能則是晶胚半徑的函數(shù)[33]。反應(yīng)相從光滑的顆粒表面溶解也同樣需要額外的自由能來形成最初的溶蝕坑。圖4為鈉長石的溶解速率曲線,在稀溶液狀態(tài)(即高度欠飽和狀態(tài))時,例如礦物相與液相接觸的瞬間,液相中可溶解礦物相的濃度是極低的,此時礦物相的溶解速率很高,因?yàn)榍凤柡腿芤禾峁┑淖杂赡芸梢栽诘V物相表面形成最初的溶蝕坑[6-7,26-27,29]。而液相濃度高于某一閾值后,溶解速率急劇降低,因?yàn)橐合嗳鄙僮銐虻淖杂赡茉诘V物相表面繼續(xù)溶蝕。而溶蝕晶體現(xiàn)有缺陷處所需的自由能低于溶蝕光滑的顆粒表面,因而溶解得以繼續(xù)進(jìn)行,但溶解速率下降。一般來說,形成溶蝕坑所需的自由能與伯氏矢量 (Burger’s vector) 有關(guān)。由于膠凝材料的晶體結(jié)構(gòu)特點(diǎn)以及位錯特性,晶體缺陷越多,誘導(dǎo)期越短[34-36]。Juilland等[22]使用SEM觀察了不同條件下Alite顆粒的水化表面,發(fā)現(xiàn)在水中水化的Alite顆粒表面有明顯的溶蝕坑,而在飽和CH溶液中水化的樣品則保持了光滑的顆粒表面。該結(jié)果表明溶解假說可以為文獻(xiàn)中的大量結(jié)果提供合理的解釋,而且該過程也有試驗(yàn)結(jié)果的支持[37]。

圖4 鈉長石的溶解速率曲線[30]Fig.4 Dissolution rate curve of albite[30]
3 C3S體系的早期水化
3.1 C3S體系再次水化加速的原因
Skalny[15]列出了四種可能導(dǎo)致C3S體系再次水化加速的誘因,分別是C-S-H的形核和生長,穩(wěn)定態(tài)C-S-H的生長,C-S-H(m)的破裂,以及CH的形核。
Bullard[38]使用模型模擬C3S體系水化進(jìn)程中微觀結(jié)構(gòu)的發(fā)展以及溶液組成的變化,發(fā)現(xiàn)C-S-H的形核發(fā)生在C3S水化開始的前幾分鐘內(nèi)。而且Bellmann等[18]的工作也說明C-S-H的生成先于C3S水化加速期的出現(xiàn),因此,僅C-S-H的形核很難成為促進(jìn)C3S體系再次水化加速的必要條件。
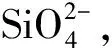
在C3S體系水化進(jìn)程中,CH以及C-S-H晶體的沉淀和生長幾乎同時進(jìn)行。由于摻入CH可以延緩C3S體系的水化作用,因此CH的析出也曾被認(rèn)為是C3S水化速率再次上升的原因之一。但是,在C3S水化樣中摻入CH晶種并不會加速C3S體系的水化[39],甚至可能延緩加速期的出現(xiàn)[40]。同樣,相比在水中進(jìn)行C3S水化,CH溶液可以延緩C3S體系的水化進(jìn)程[22]。因此,摻入少量的CH晶體并不能起到提供晶核的作用,因?yàn)镃H晶體遇到水時會迅速溶解,溶液關(guān)于反應(yīng)相的欠飽和度降低,從而降低C3S的溶解速率[22,41]。即使CH在溶液中快速飽和,由欠飽和度降低引起的反應(yīng)相溶解速率下降將延緩C3S水化加速期的出現(xiàn)。隨著液固比增加,CH的溶解度大于C-S-H,所以需要溶解更多C3S來析出CH。在此條件下,CH的沉淀是C3S加速溶解的結(jié)果,而不是導(dǎo)致其加速溶解的原因[42]。

3.2 C-S-H的生長與結(jié)構(gòu)
目前,學(xué)者們確信C-S-H的生長與C3S體系的水化有關(guān),而C-S-H的生長影響了所觀察到的C-S-H結(jié)構(gòu)。關(guān)于C-S-H結(jié)構(gòu)的生長,存在以下兩種理論,即納米顆粒聚集理論 (aggregation of nanoparticles)[45-49],以及大而有缺陷的硅酸鹽層理論 (large and defective sheets of silicate)[50]。
Gartner[50]提出了關(guān)于C-S-H的分支片狀 (branching sheets) 生長機(jī)制,即硅氧四面體沿著二維方向形成聚集體,并且在這些層狀硅酸鹽結(jié)構(gòu)中插入Ca2+以及—OH形成類似托貝莫來石 (tobermorite) 或六水硅鈣石 (jennite) 的結(jié)構(gòu)。C-S-H在形核后以該種方式生長,并且隨著生長的進(jìn)行,這些層狀結(jié)構(gòu)在納米尺度上形成“短程有序”的原子排列。然而,隨著層狀結(jié)構(gòu)的不斷生長,由插層導(dǎo)致的點(diǎn)缺陷或線缺陷不斷增多,這些生長缺陷引起晶體結(jié)構(gòu)扭曲從而導(dǎo)致原子排列的“長程無序”,因此C-S-H在微觀尺度上表現(xiàn)為晶體結(jié)構(gòu)的無序化,也被稱作C-S-H凝膠。
另一種C-S-H結(jié)構(gòu)生長的理論是關(guān)于C-S-H納米顆粒的聚集理論。該理論認(rèn)為C-S-H固體顆粒僅能生長到幾納米便長時間保持穩(wěn)定。然而,現(xiàn)有的C-S-H納米顆粒可以促使新的顆粒在其表面形核,或者先前在溶液中形核的C-S-H顆粒在其表面聚集[44]。但是,新的形核過程要比在現(xiàn)有的C-S-H晶核表面繼續(xù)生長所需的自由能更高 (即過飽和度更高),而且一些試驗(yàn)和模擬中,在高過飽和度狀態(tài)下,只有水化最初的幾分鐘內(nèi)可以出現(xiàn)C-S-H形核[21,26,41]。所以該理論可能存在一定的局限性。

圖5 不同摻量下體系的 水化放熱曲線[51]Fig.5 Heat flow curves of system hydration with different dosages of

5 結(jié) 語
猜你喜歡
小讀者(2021年2期)2021-03-29 05:03:48
新世紀(jì)智能(數(shù)學(xué)備考)(2020年11期)2021-01-04 00:38:16
瘋狂英語·新悅讀(2019年11期)2019-12-18 05:14:16
華人時刊(2019年13期)2019-11-17 14:59:54
中國外匯(2019年17期)2019-11-16 09:31:14
NBA特刊(2018年21期)2018-11-24 02:48:04
文苑(2018年22期)2018-11-19 02:54:14
紅領(lǐng)巾·萌芽(2016年1期)2016-09-10 07:22:44
現(xiàn)代企業(yè)(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
