手性環戊二烯基配體參與的不對稱催化C—H活化/環化反應研究進展
2021-08-17 00:58:16嚴知靜程澤生楊子瀟陳欣怡
合成化學 2021年7期
關鍵詞:催化劑
嚴知靜, 程澤生, 楊子瀟, 陳欣怡, 董 琳*
(1. 四川大學 a. 華西藥學院; b. 生物治療國家重點實驗室,四川 成都 610041)
自從2012年Gramer課題組[1]基于環戊二烯配體發展了一系列的BINOL型C2軸手性銠金屬催化劑以來,在過去的十幾年中,含有環戊二烯基配位的過渡金屬復合物催化的C—H鍵官能團化反應研究取得了顯著的進展[2-6]。目前人們已經制備出大量環戊二烯基配位的過渡金屬復合物催化劑來催化各類環化反應構造不同結構的手性環狀化合物[7-8],如通過[4+1]環化反應構造異吲哚啉酮類和內酯類化合物,通過[4+2]環化反應構造異喹啉酮類和內酰胺類化合物,以及[2+2]環化反應、螺環化反應等等。
近年來手性環戊二烯基配位的過渡金屬絡合物在C—H活化取得了較好的成績,已成為有機合成路線、高效簡便、選擇性好、原子經濟性高等優點。這對于以這類環狀化合物結構為主要原料的藥物催化合成、工業化大規模生產投放有著重要意義。
然而在實際的藥物合成生產中,關于環狀化合物類原料及前體藥物催化合成中的對映選擇性的研究仍然較少。因此,本文對近年來關于手性環戊二烯基配體催化的環化反應研究進行了歸納、總結,以期能幫助此類環狀化合物及相關藥物化學合成的研究,特別是對各類手性環戊二烯基配位過渡金屬催化劑的催化原理及設計合成思路給予新的啟發。
1 手性金屬催化劑參與的[4+1]環化反應
1.1 手性異吲哚啉酮類化合物的合成
2019年,Gramer等[9]報道了手性CpxRhIII催化的丙烯酰胺類化合物的對映選擇性C—H活化/[4+1]環化反應,以構建一系列含有手性季碳中心的ɑ,β-不飽和γ-內酰胺類化合物(Scheme 1),其對映選擇性高達97/3。
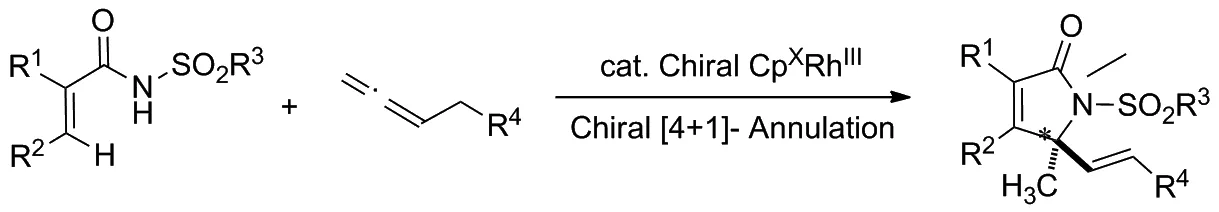
Scheme 1
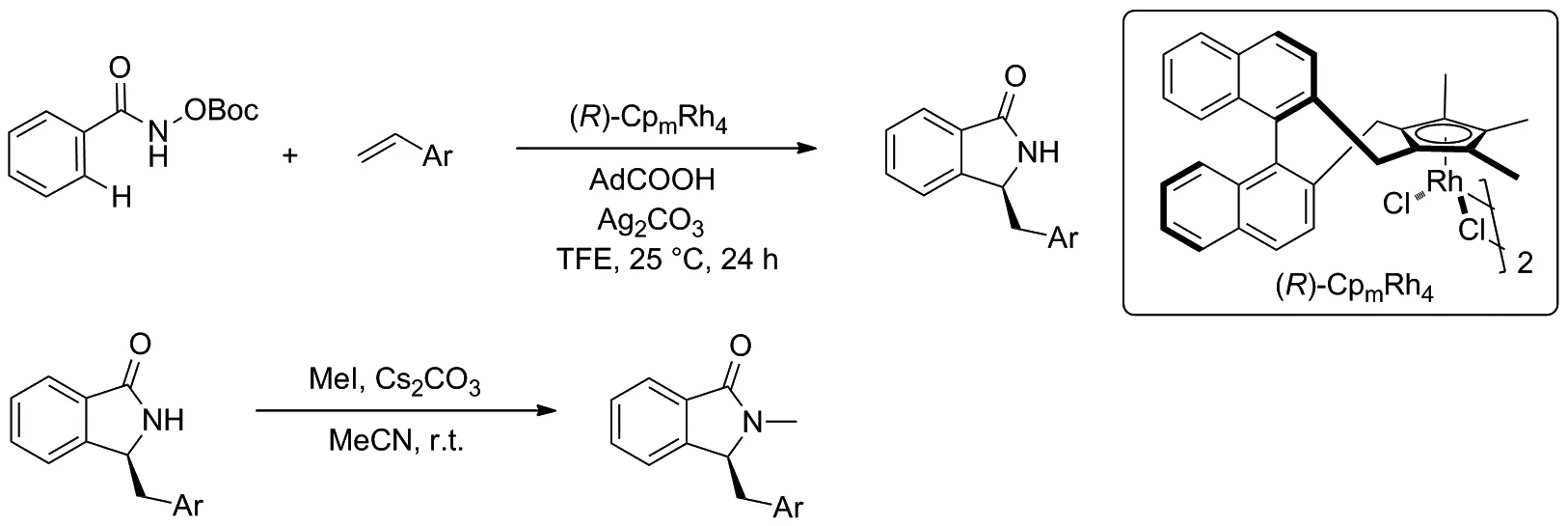
Scheme 2
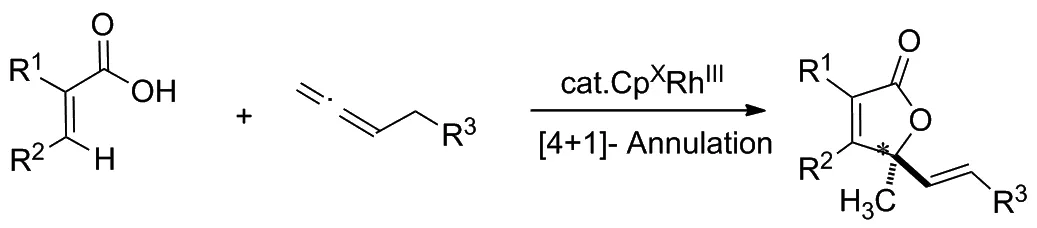
Scheme 3
2020年,You等[10]實現了CpxRhIII催化的苯甲羥肟酸衍生物與苯乙烯之間的環化反應(Scheme 2)。該反應在溫和的反應條件下,經過C—H活化/[4+1]環化串聯反應,以84%的產率得到3-取代異吲哚酮化合物,對映選擇性高達96/4。 然后,標準產物在碳酸銫存在下,與碘甲烷進行取代反應,得到N-甲基取代產物的產率為96%,而對映選擇性沒有明顯降低。
1.2 手性γ-內酯類化合物的合成
2020年,Gramer等[11]以部分氫化的聯二萘酚為基本骨架,設計了一種新穎的手性Cpx配體。該反應的底物適用性廣泛,通過不同結構的丙烯酸類衍生物和聯烯類化合物反應,得到了一系列具有烯丙基立體中心的不飽和γ-內酯類化合物,其對映選擇性可高達99/1(Scheme 3)。
2 手性金屬催化劑參與的[4+2]環化反應
2.1 手性異喹啉酮類化合物的合成
在以往的C—H活化/[4+2]環化的相關報道中,手性化合物的合成往往采用分子間合成的方式,而分子內的[4+2]環化合成更具有挑戰性。2018年,Antonchick和Waldmann課題組[12]以不同取代的哌啶環結合環戊二烯基而成的銠金屬為催化劑,探究了N-氧烷基苯甲酰胺類化合物與炔烴的分子內[4+2]環化反應,同時,利用底物本身的芳(雜)環結構合成出結構新穎的軸手性異喹啉酮類化合物(Scheme 4)。該反應具有較高的產率與對映選擇性,且在對底物進行不同的結構修飾時,仍可以保持較高的對映選擇性。
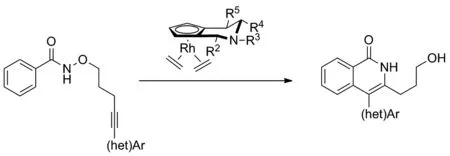
Scheme 4
Scheme 5
Scheme 6
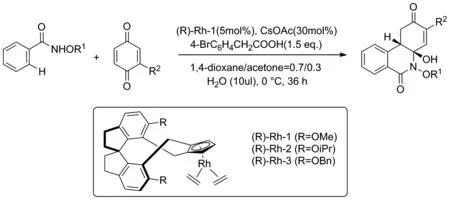
2020年,Gou和Wang課題組[13]以N-甲氧基苯甲酰胺類化合物與苯醌為反應原料,合成一系列的手性異喹啉酮類衍生物(Scheme 5)。在反應中,苯醌發揮了雙重作用,一方面將催化劑CpRhII(C2H4)2原位氧化成CpRhIII催化反應進行,另一方面參與反應中間體的形成,通過自身與催化劑發生配位推動反應的進行,從而幫助完成[4+2]分子間環化反應。該反應通過螺環化的手性銠金屬催化劑進行立體選擇性的合成,具有很高的產率與對映選擇性。
2.2 手性α,β-不飽和內酰胺類化合物的合成
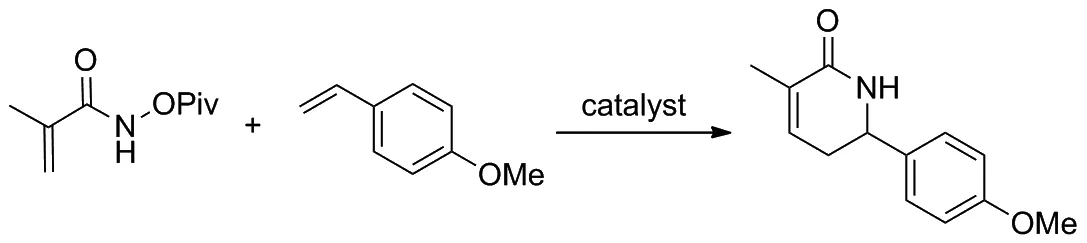
2019年,McNaughton和Rovis課題組[14]提出利用人工金屬酶進行不對稱催化C—H活化和[4+2]環化反應來合成手性α,β-不飽和內酰胺類化合物,并以此作為藥物合成中間體構造哌啶(Scheme 6)。該反應以甲基丙烯酰胺與苯乙烯及其衍生物為反應底物,獲得的產品可以衍生為具有良好對映選擇性的哌啶。課題組還發現單體鏈霉親和素相較于四聚體親和素具有更好的催化活性,其所得產物也具有更高的產率和更好的對映選擇性。
2.3 手性磷類化合物的合成
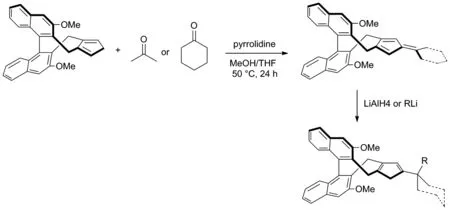
2018年,Sun 和Cramer等[15]開發了一系列三取代的手性環戊二烯配體并利用該類催化劑進行手性含膦化合物的合成(Scheme 7)。實驗證明了雙取代手性環戊二烯配體骨架可以與酮縮合成相應的富烯[16],并在LiAlH4或RLi的還原下生成相應的連有較大取代基的三取代手性環戊二烯的催化劑配體。
Scheme 7
Scheme 8
Scheme 9
Scheme 10
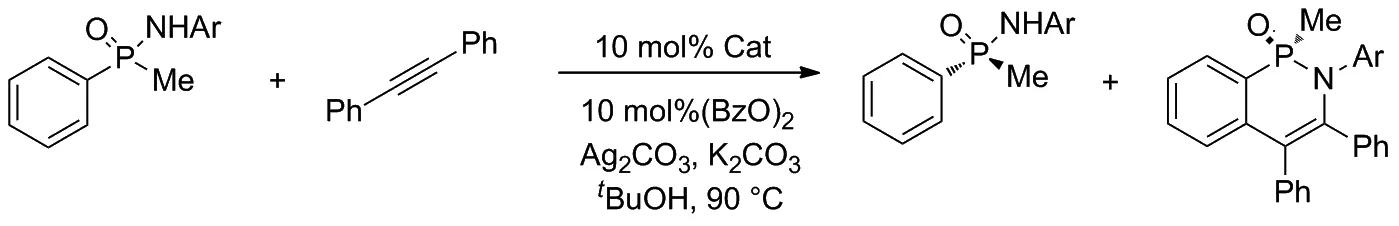
隨后的實驗證明,在環戊二烯中心位置引入體積較大的第三取代基可以顯著提高動力學拆分的選擇性,并通過動力學拆分提供手性膦化合物。實驗還使用不同類型的催化劑參與外消旋苯基甲基次膦酰胺和二苯乙炔的反應,并統計了不同催化條件下所得產物的產率,驗證了Cpx三取代催化劑優越的選擇性(Scheme 8)。
2. 4 手性磺酰亞胺類化合物的合成
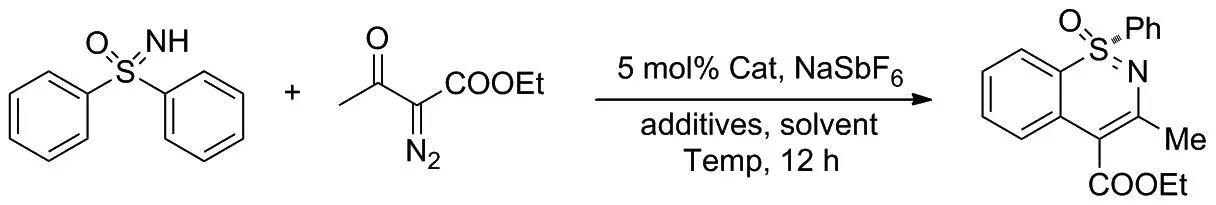
磺胺嘧啶中的立體中心:硫原子是一種潛在的手性藥物結構單元。2018年, Sun和Cramer[17]提出了一種直接催化合成含有手性硫原子的1,2 -苯并噻嗪類化合物的方法(Scheme 9)。
Scheme 11
Scheme 12
Scheme 13
該課題組在手性環戊二烯基銠(III)催化劑與適當的酸類添加劑的作用下,以二苯亞砜和重氮乙酰乙酸乙酯為模板底物,合成手性的1,2-苯并噻嗪類化合物,得到良好的收率和對映選擇性。同時,他們還發現該轉化過程對不同取代修飾的磺基亞胺均具有較高的對映選擇性,而使用手性酸類添加劑能產生協同作用并提高反應的選擇性。
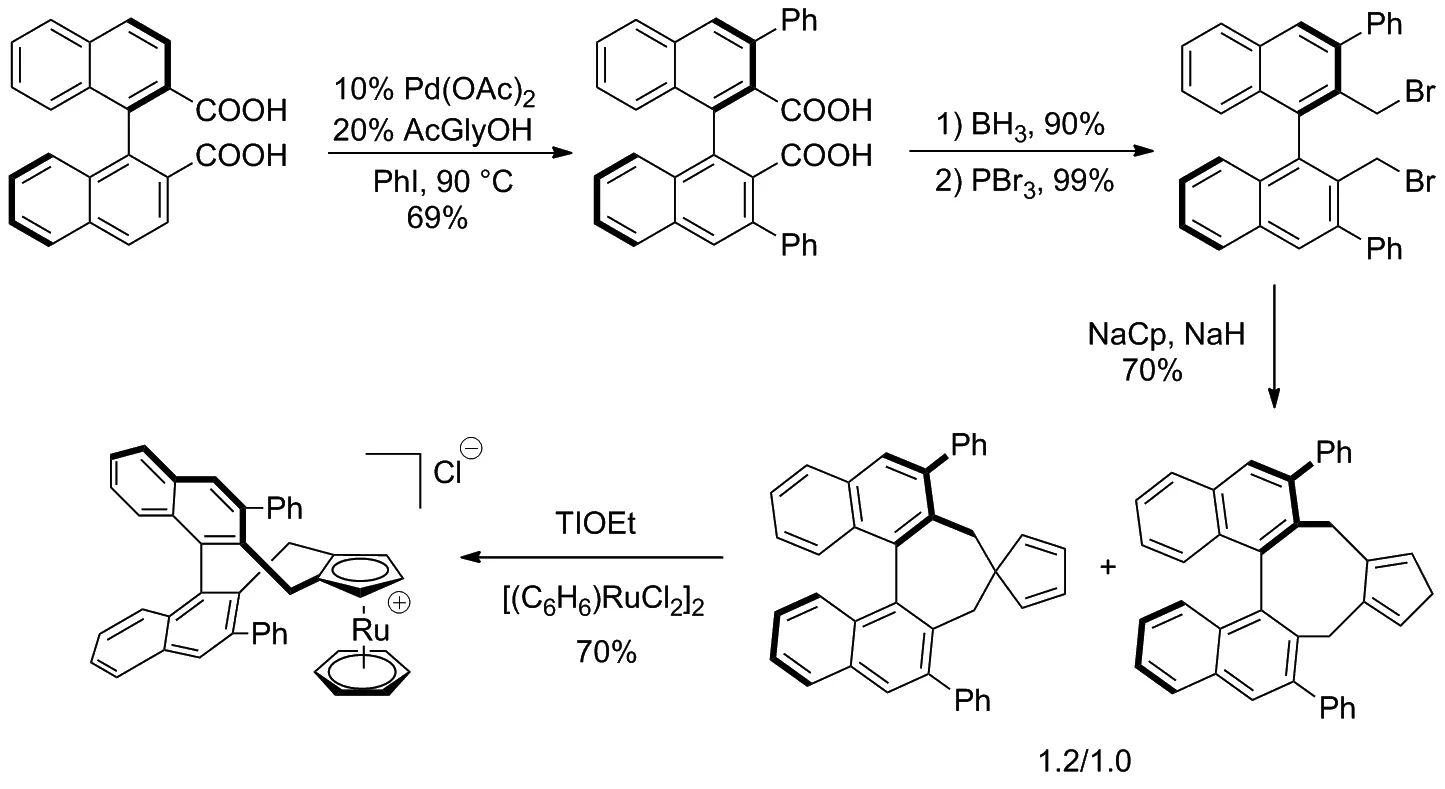
2017年Cramer等[18]設計了一種手性CpRuIICl絡合物并縮短了該催化劑的合成路線(Scheme 10),用適量PdII催化二羧酸[19]與碘苯的偶聯反應(雙羧酸作為導向基團),進行鄰位活化[20]從而獲得雙芳基產物,經還原和取代后得到了溴代產物,再通過Cp基團環化和絡合生成穩定的手性CpRuII配合物。
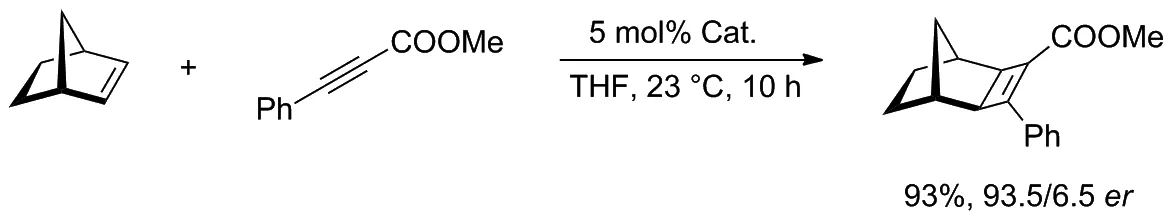
在以降冰片烯和炔烴為原料生成環丁烯的實驗中(Scheme 11),該類催化劑對生成[2+2]-環加物表現出優異的反應活性和對映選擇性,并降低了催化劑的負載量。催化劑中的氯離子具有很強的反離子作用,可將相應的非選擇性陽離子絡合物轉化為具有高度對映選擇性的中間體。
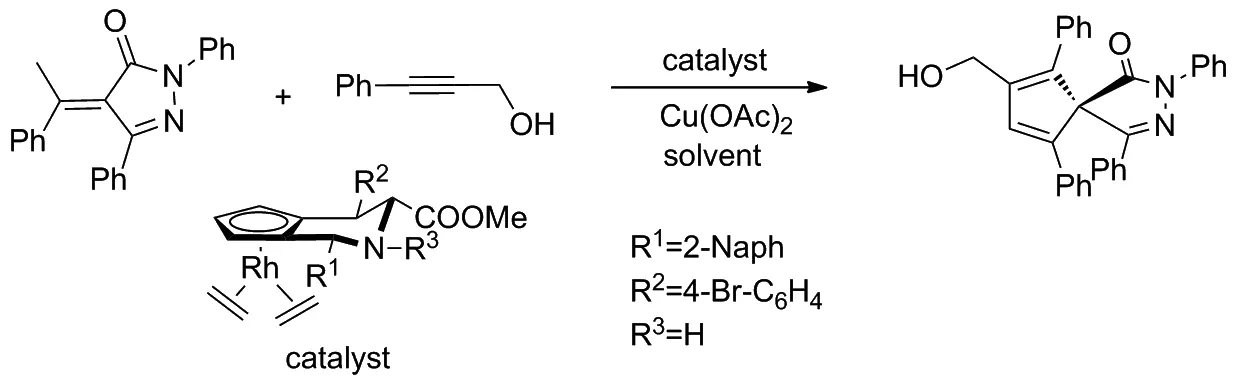
2019年,Waldmann等[21]以 CpxRhIII為催化劑,使用α-芳基吡唑啉酮和不對稱芳基炔進行對映選擇性環化反應,通過C(sp3)—H活化獲得一系列包含全碳四元中心的螺并吡唑啉酮(Scheme 12)。該反應產率高、產物對映選擇性好,可以在溫和的反應條件下獲得結構多樣的產品。
5 手性金屬催化的軸手性化合物的合成
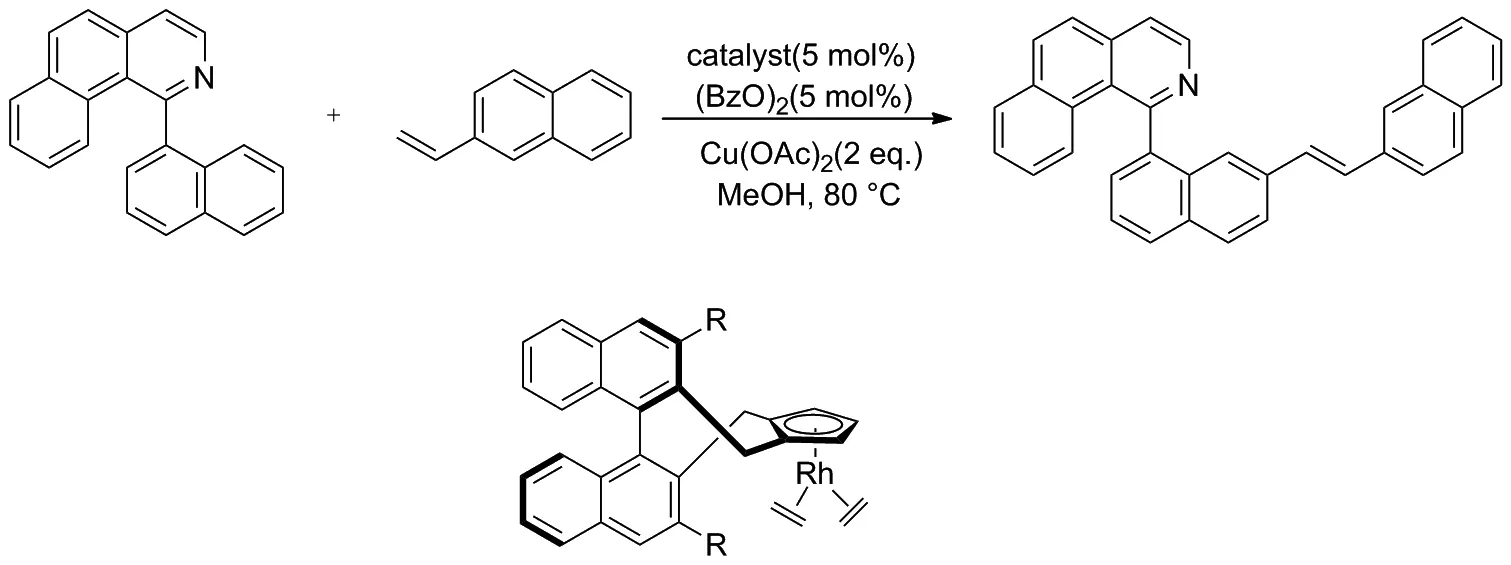
2014年, Zheng和 You等[22]報道了1-(萘-1-基)苯并[h]異喹啉和2-乙烯基萘聯芳基的對映選擇性烯基化反應(Scheme 13)。該反應使用手性CpRhⅢ催化劑,通過聯芳基化合物的直接C—H鍵烯基化合成了新型軸手性聯芳基骨架,并取得了良好的收率和對映選擇性。
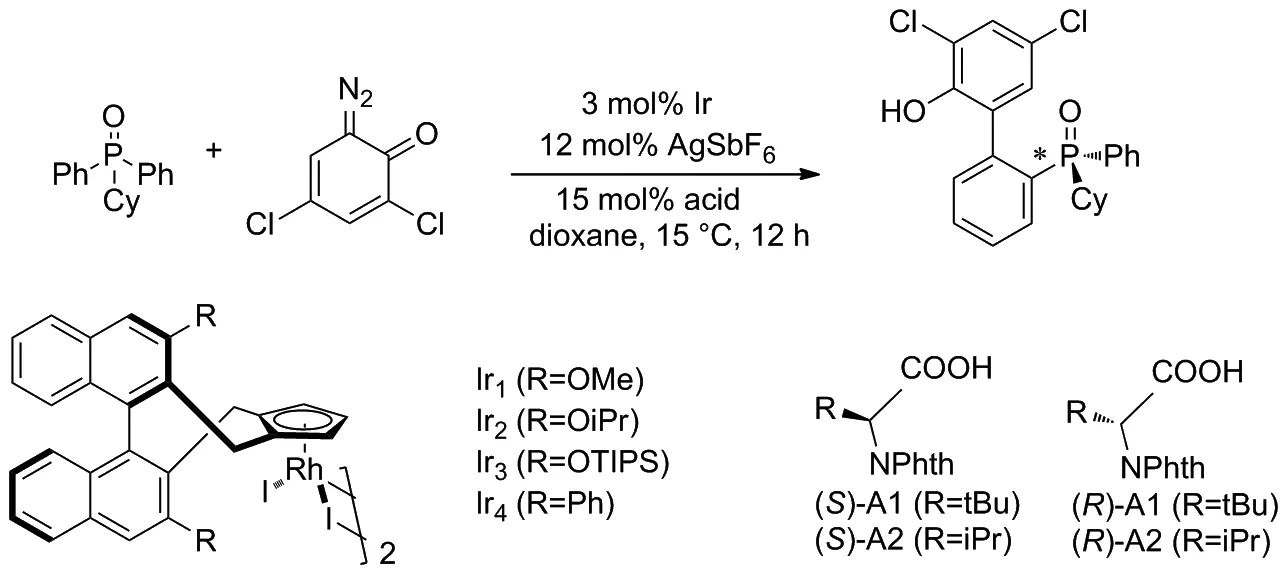
2018年,Cramer等[23]以二苯基環己基氧化膦和二疊氮化醌為原料,在IrIII催化劑的催化下通過對映選擇性C—H芳基化反應獲得了軸手性聯芳基膦氧化物(Scheme 14)。證明了絡合手性Cpx配體與帶有亮氨酸衍生的對羥基苯磺酸(S)-甲酸酯(對苯二甲酸酯基)基團的IrIII催化劑可以帶來良好的對映選擇性和收率。該技術適用于在膦上具有手性的雙芳基膦氧化物,以及對立體選擇性要求高的軸向手性聯芳基骨架的構建。
Scheme 14
本文總結了近年來關于手性環戊二烯基配體參與的環化反應的研究進展,為手性環戊二烯基配位的過渡金屬的設計合成及催化應用提供了新的思路。同時,此類催化劑具有能夠縮短合成路線、高效簡便、選擇性好、原子經濟性高等優點,對以這類環狀化合物結構為主要原料的藥物催化合成、工業化大規模生產投放有著重要意義。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
