
錳基氧化物活化過硫酸鹽降解水中有機污染物的研究進展
2021-08-23 10:29:42王一凡李小蝶侯美茹王兆慧
環境科學研究 2021年8期
王一凡, 李小蝶, 侯美茹, 王兆慧,2,3*
1.華東師范大學生態與環境科學學院, 上海市城市化生態過程與生態恢復重點實驗室, 上海 200241 2.上海有機固廢生物轉化工程技術研究中心, 上海 200241 3.自然資源部大都市區國土空間生態修復工程技術創新中心, 上海 200062
隨著人類大規模的生產活動,生活污水和工業廢水排放量的增加,水資源污染問題日益嚴重. 水體中的污染物主要包括重金屬、難降解的有機物、植物營養物、放射性物質、石油類、病原體、懸浮物等,這些污染物由于自身特征與含水介質的作用,易從地表水源遷移進入地下水中造成嚴重污染,而且很難通過常規的廢水處理方法有效去除[1].
錳基催化劑具有晶型結構及性質多變、比表面積大、毒性低、自然豐度高、環境友好[10]、成本低且可大規模制備等優勢,作為水處理中SR-AOPs的催化材料,可用于活化PMS/PDS降解水中有機污染物. Mn(Ⅱ、Ⅲ、Ⅳ和Ⅶ)氧化物因其氧化態和結構具有多樣性[11],對于過硫酸鹽的活化效率亦存在較大差異. 因此,深入理解錳氧化物的類型結構和作用原理,對其用于活化PS降解污染物具有重要的理論和現實意義. 鑒于此,該文主要對錳基氧化物活化PS降解水中有機污染物的應用研究進行了綜述. 概述了錳基氧化物的類型、結構特征、合成方法及影響反應活性的因素,針對各種錳基氧化物活化PS的反應機理進行了系統的闡述,最后提出建議和研究展望.
1 錳基氧化物的類型、結構特征及合成方法
1.1 類型與結構特征
錳氧化物在自然界中廣泛存在,富含于海洋和陸地環境中. 作為天然存在的強氧化劑之一,廣泛參與環境中各種化合物的氧化還原反應. 由于錳的價態十分豐富,因此其氧化物的存在形式也較為多樣. 常見的錳氧化物有幾十種,如軟錳礦(MnO2)、褐錳礦(Mn2O3)、硬錳礦(mMnO·MnO2·nH2O)、水錳礦[MnO2·Mn(OH)2]等. 錳氧化物隨氧化價態的升高,堿性降低,酸性增強. 同時,錳元素的氧化價態越高,其氧化物的催化活性越強. 錳氧化物通常具有較高的化學反應活性,可以通過吸附、氧化分解等形式對有機物的轉化產生影響.
錳氧化物的晶體類型和形態結構是影響其催化反應性能的重要因素. 二氧化錳的晶體結構分為3類,即一維隧道結構、二維層狀結構及三維網狀結構,主要存在的晶體類型包括α-MnO2、β-MnO2、γ-MnO2、λ-MnO2、δ-MnO2等. 錳氧化物基本結構是由1個Mn原子和6個O原子配位形成的六方密堆積結構及立方密堆積結構,即[MnO6]八面體與鄰近的八面體會共用棱與角頂,從而形成復雜的網格結構. 不同形態結構的錳氧化物,在化學反應過程中表現出不同的催化反應性能.
1.2 合成方法
常見合成錳基氧化物的方法有水熱法[12]、溶劑熱法[13]、溶膠凝膠法[14]、沉淀法[15]、微乳液法[16]等. 水熱法是合成錳基材料的經典方法,可以從含有不同摩爾比的MnCl2和KMnO4的體系中制備α-MnO2、β-MnO2、γ-MnO2[17]. 3種晶體結構的MnO2在低溫條件下均具有較高的催化反應活性. Xiao等[18]采用改進的水熱法工藝,通過還原KMnO4,改變前驅物的濃度及水熱的溫度來制備3D分層結構的MnO2催化劑. Cao等[19]利用模板法和水熱法制備4種不同形態(空心球、空心立方體、納米棒、海膽)的Mn2O3,其中空心球和空心立方體結構的Mn2O3具有較強的穩定性及催化活性. Tian等[20]以高錳酸鉀和氯化氫為原料,利用水熱法在不同溫度下分別合成不同形狀(納米管、納米棒和納米顆粒)的MnO2,其中納米棒的MnO2具有較高的催化性能. Atiqueullah等[21]在含有KMnO4和甘油的體系中,利用溶膠-凝膠法制備Mn5O8(單斜晶和納米棒結構)和Mn2O3的復合納米顆粒. Meng等[22]在無任何催化劑或模板劑的情況下,利用低溫水熱法合成γ-MnOOH納米棒,并以其為前驅物在高溫煅燒下制備出β-MnO2納米棒以及MnO、Mn2O3、Mn3O4納米粒子.
2 影響反應活性的因素
錳基氧化物活化PDS或PMS受自身物理化學性質、外部反應條件的影響,包括反應溫度、溶液pH、晶型結構、共存離子、污染物初始濃度、催化劑的用量等[23].
2.1 共存陰離子
水環境中通常存在大量的無機陰離子,如碳酸氫根(HCO3-)、硝酸根(NO3-)、亞硝酸根(NO2-)、氯離子(Cl-)、CO32-、硫酸根(SO42-)等. 在PMS氧化體系中這些無機陰離子可消耗或進一步產生活性自由基[24]. 例如,NO2-與SO4·-和·OH易發生反應〔見式(1)~(2)〕,與污染物競爭活性自由基,因此表現出比其他無機陰離子更強的抑制效果.
417 Progress in the detection of macular ganglion cell complex thickness
SO4·-+NO2-→NO2·+SO42-,k=8.8×108L/(mol·s)
(1)
·OH+NO2-→NO2·+OH-,k=8.0×109L/(mol·s)
(2)
共存離子SO42-和Cl-對MnO2/NF/PMS體系AO7降解動力學呈現不同的影響規律. SO42-存在的情況下,AO7的降解率未受明顯影響,然而隨著Cl-的添加量逐漸增多,染料降解呈現顯著的加速趨勢. HCO3-和CO32-能以不同速率與SO4·-和·OH反應,因此二者常作為自由基的主要清除劑,抑制有機污染物的降解[25]. 若環境中存在過量的天然有機質(NOM), 也會消耗SO4·-或·OH從而降低降解效率[26].
氯化物和碳酸鹽對不同的活化過程表現出迥異的影響規律[27]. 在PMS/Mn3O4-MnO2體系共存陰離子會抑制污染物環丙沙星(CIP)的降解,抑制作用依次表現為CO32->HCO3->NO3->SO42-,而Cl-起促進降解的作用[28]. 原因可能在于HCO3-與CO32-將SO4·-和·OH 清除,并產生了氧化能力較弱的碳酸鹽自由基[29]. Cl-直接與PMS發生反應生成活性氯(HClO/Cl2)或者Cl-與SO4·-和·OH反應生成氯自由基,對基于PMS氧化體系的影響程度受pH和Cl-濃度控制. Deng等[30]研究表明HCO3-和CO32-均抑制卡馬西平(CBZ)的降解速率. 由于CO32-與SO4·-的反應速率約是HCO3-的4倍,因此CO32-相較于HCO3-對CBZ降解的抑制作用更明顯. 此外,陰離子還可以通過競爭吸附作用影響污染物的降解效率. 一方面,通過占據催化劑表面的活性位點抑制催化劑與氧化劑的接觸,影響自由基的生成效率;另一方面,較高濃度的陰離子會抑制污染物在錳氧化物表面的吸附.
2.2 溶液pH
溶液pH會改變錳基材料表面電荷分布,影響材料與PMS/PDS之間的相互作用及反應活性[31]. Jo等[32]研究氧化鐵固定化MnO2復合材料活化PS時發現,pH為9.0時四氯化碳和苯的降解率最高. 但當pH較低時錳氧化物會溶解,Mn(Ⅱ)釋放改變了Mn基材料的表面結構. 若存在一定量的質子與PMS/PDS 結合,也會干擾并抑制Mn基材料的催化反應活性[33]. 然而,Xiao等[34]發現pH為3.0~7.0時從含錳廢水中合成納米結構MnO2對苯酚的降解率較高. 原因在于:PMS在堿性條件下不穩定,易分解成O2和SO42-;酸性條件能夠促進SO4·-和·OH的生成,且該條件下的自由基具備較高的氧化還原電位. 因此,隨著初始pH的增加,污染物的去除率逐漸降低[35]. MnO2/UV/PMS體系降解4-氯苯酚也具有上述相同的反應趨勢[36],即酸性條件下污染物的降解效率較高.
2.3 反應溫度
反應溫度是影響錳基氧化物活化PMS/PDS效率重要因素之一. Mn基氧化物活化PMS/PDS過程中,有機污染物的降解速率與溫度呈正相關[37-38]. 原因在于升高溫度,PMS/PDS自身的熱活化、Mn基氧化物活化和自由基降解污染物的反應都會加速. 但反應溫度并非越高越好,要根據催化劑類型、氧化劑穩定性和處理成本選擇適宜的反應溫度.
式(3)表示速率常數[k,L/(mol·s)]與溫度之間的相關性,可用于計算活化能.
lnk=lnA-Ea/RT
(3)
式中:A為頻率因子,L/(mol·s);Ea為活化能,J/mol;R為通用氣體常數,J/(mol·K);T為絕對溫度,K. 不同錳氧化物降解同一種污染物,會計算得出不同的活化能. 由于錳氧化物活化PMS/PDS 和SO4·-氧化降解苯酚等多個反應都受溫度影響,因此計算得到的Ea其實為包含多個基元反應的表觀活化能. 不同體系之間不宜采用Ea來比較錳氧化物的催化性能.
2.4 晶型結構
不同形態結構的錳基氧化物活化PMS時展現出不同的催化反應活性,例如,結晶態的α-MnO2具有較高的苯酚降解率[39]. 不同形狀的Mn2O3晶體催化苯酚的降解速率依次表現為Mn2O3(正立方體)>Mn2O3(正八面體)>Mn2O3(八面體截斷)[40]. Wang等[41]制備的一維納米α-MnO2的催化活性表現為α-MnO2(納米線)>α-MnO2(納米棒)>α-MnO2(納米管). 不同結晶相的一維MnO2納米顆粒(α-MnO2納米線、β-MnO2納米棒、γ-MnO2納米纖維)催化活性大小為α-MnO2>γ-MnO2>β-MnO2. α-MnO2的反應性和穩定性最高[42]. Huang等[43]建立了4種不同MnO2結構的催化反應活性與物理化學性質之間的線性關系,得出反應活性順序依次為α-MnO2>γ-MnO2>β-MnO2>δ-MnO2. 此外,催化反應性還取決于Mn的氧化態,反應趨勢表現為Mn2O3>MnO>Mn3O4>MnO2[44].
3 錳基氧化物活化PMS的機制
3.1 MnO2及其復合型氧化物活化PMS
MnO2活化PMS產生的SO4·-和·OH作為主要的氧化性物種能有效去除苯酚、BPA和有機合成染料等污染物〔見式(4)~(7)〕.
HSO5-+2MnO2→SO5·-+OH-+Mn2O3
(4)
HSO5-+Mn2O3→SO4·-+H++2MnO2
(5)
SO4·-+H2O→·OH+H++SO42-
(6)
SO4·-+OH-→·OH+SO42-
(7)

(8)
(9)
(10)
(11)
RhB+SO4·-→CO2+H2O+SO42-
(12)
除RhB外,BPA在PMS-MnO2體系氧化降解存在2種反應過程:一是MnO2的直接氧化[48-49],在酸性條件尤為明顯;二是催化氧化,α-MnO2、β-MnO2以及γ-MnO2活化PMS均可生成SO4·-和·OH和1O2. Wang等[50]通過乙醇(EtOH)和叔丁醇(TBA)2種清除劑的自由基淬滅試驗,證實SO4·-在活化降解過程中起主導作用. Dong等[51]揭示了β-MnO2納米線具有良好的可分離性及顯著的催化降解作用. Sui等[52]證明α-MnO2和β-MnO2單晶體納米結構顆粒在去除亞甲基藍的類芬頓反應中表現出優異的催化性能[53].
除上述單一錳氧化物外,α-MnO2負載Co3O4納米顆粒可以有效非均相活化PMS降解水溶液苯酚〔見圖1(a)〕[54]. 僅采用Co3O4對PMS進行活化需要使用專門制備的惰性載體,且反應活性過分依賴于載體. 而Co3O4與MnO2的復合對PMS的活化和苯酚的降解具有顯著的協同作用. Co3O4/MnO2具有較高的氧化還原電位,能夠加快SO4·-的生成,提高苯酚的降解速率. 因此,SO4·-的生成效率取決于Co3O4與MnO2的氧化還原反應.
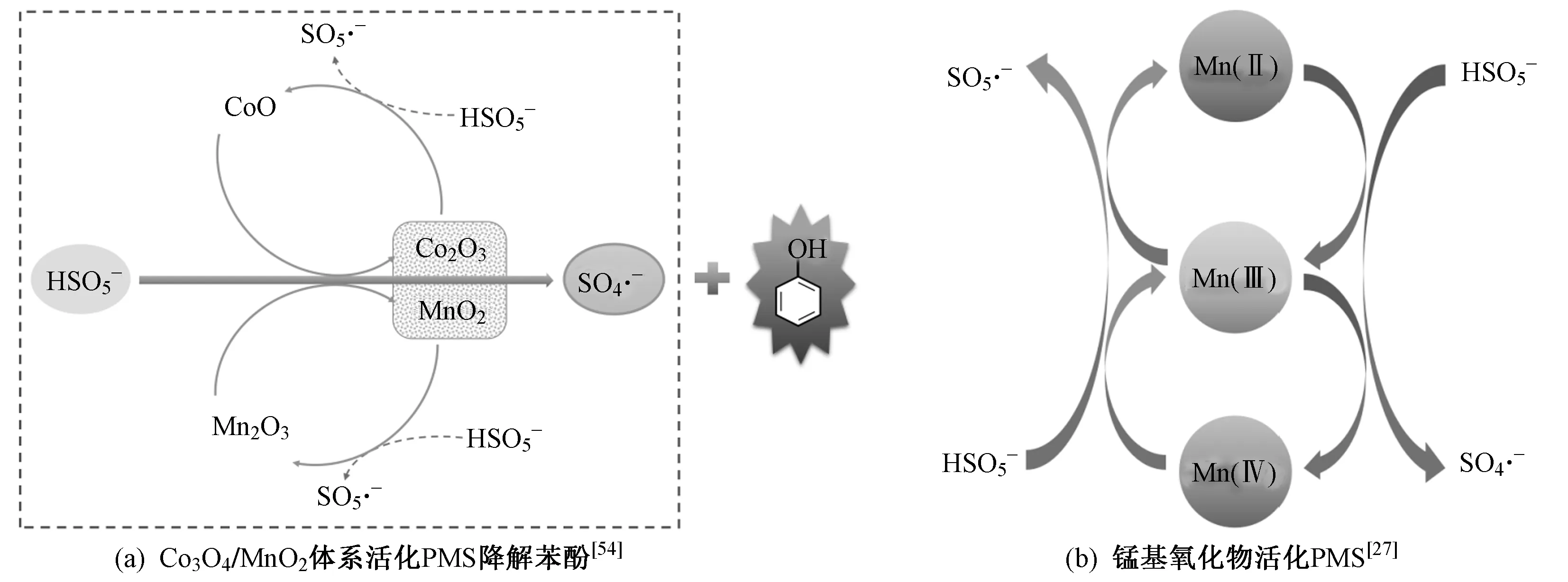
圖1 錳基氧化物活化PMS的反應機理Fig.1 Reaction mechanism of PMS activation by manganese-based oxides
此外,淬滅試驗和電子自旋共振(ESR)數據證明,除自由基機理外,非晶態的MnO2可通過非自由基機理活化PMS,生成PMS-MnO2絡合物降解有機污染物[55]. 在非晶態MnO2和PMS之間形成的活性絡合物可以介導BPA和PMS之間的直接電子轉移,選擇性地氧化酚類化合物. 因此,非晶態的MnO2/PMS 體系在有機化合物選擇性氧化方面具有較好的應用前景.
3.2 MnOx及其復合型氧化物活化PMS
除MnO2外,其他錳基氧化物(MnO、Mn2O3、Mn3O4等)也可用于活化PMS,且相較于一元氧化物具有更好的催化效果以及高穩定性、低成本、多價態、高催化活性等優勢,其反應機理〔見式(13)~(15)〕如下:
HSO5-+2MnO→SO4·-+H++Mn2O3
(13)
HSO5-+2Mn3O4→SO4·-+H++3Mn2O3
(14)
HSO5-+Mn2O3→SO4·-+H++2MnO2
(15)
近年的研究表明,具有混合Mn價態和不同納米結構、形狀等的錳氧化物復合材料,或與其他金屬元素材料形成的新型復合材料可增強催化反應活性. Saputra等[56]合成的Mn3O4和Co3O4納米顆粒活化降解效果顯著,反應活性表現為Mn3O4>Co3O4>Fe3O4. 但金屬離子Co2+具有毒性,對人體健康的危害較為嚴重,不推薦使用.
如上所述,錳氧化物催化劑在催化PMS的過程中除產生自由基外還可能會生成單態氧1O2[57-58]〔見式(16)(17)〕.1O2是O2的激發態,在光化學過程中通過光誘導使能量轉移到O2而產生[59].1O2作為一種溫和的選擇性氧化劑,不易被常用的自由基捕獲劑甲醇或叔丁醇等淬滅[60]. 除光誘導外,還可以通過PMS的自分解、羥基自由基的重組、表面吸附的O2獲取能量[61]等方式產生1O2,如Zhou等[62-63]利用苯醌活化PMS生成1O2.
HSO5-+SO52-→HSO4-+SO42-+1O2
(16)
2·OH→1/21O2+H2O
(17)
Tian等[64]通過降解苯酚和四環素(TC)研究生物氧化錳(BioMnOx)和3D α-Mn2O3活化PMS的能力,試驗證明BioMnOx/PMS體系同時產生了SO4·-和1O2.1O2是苯酚降解的主要活性氧物種,它的產生可能僅來自PMS和BioMnOx的相互作用. 因此,相較于3D α-Mn2O3,BioMnOx表現出較高的催化活性、降解效率和穩定性. Huang等[65]合成了新型Mn1.8Fe1.2O4納米球,利用雙金屬氧化物中的2種不同金屬(Mn和Fe)陽離子之間的協同作用催化PMS降解BPA,其催化性能明顯高于Mn/Fe的單金屬氧化物. Mn(Ⅱ)和Fe(Ⅱ)向PMS提供電子,Mn(Ⅲ)提供額外的單電子活化PMS. 隨后,高價態的Mn和Fe被HSO5-還原,通過Mn和Fe氧化還原循環維持Mn1.8Fe1.2O4的催化作用〔見式(18)~(20)〕.
(18)
(19)
(20)
Sahoo等[66]曾提出Mn和Fe之間的電子轉移是二者產生協同作用的主要原因. 大多數有機污染物的多相催化降解發生在氧化物的表面,因此反應底物對活性位點的預吸附(主要是絡合效應)顯得尤為重要[67-68]. 當磷酸鹽與催化劑表面過渡金屬發生配位而使其無法裸露于表面時,Fe/Mn協同作用被抑制,與Fe配位的磷酸鹽取代了PMS,降低了降解效率. Yao等[69]證實磁性MnFe2O4納米顆粒和MnFe2O4/還原氧化石墨烯(MnFe2O4-rGO)復合物在活化PMS降解水中有機染料(甲基紫、甲基橙、亞甲基藍、橙Ⅱ和羅丹明B)時具有較高的活性和良好的穩定性. MnFe2O4納米催化劑表面上的Fe(Ⅱ)/Mn(Ⅱ)與PMS反應生成SO4·-,Fe(Ⅲ)/Mn(Ⅲ)再被PMS還原生成更多Fe(Ⅱ)/Mn(Ⅱ)〔見式(21)~(24)〕. 類似于類Fenton反應[70-71],Mn(Ⅱ)/Mn(Ⅲ)和Fe(Ⅱ)/Fe(Ⅲ) 是生成SO4·-的氧化還原對.
(21)
(22)
(23)
(24)
除雙金屬氧化物之外,Zhang等[72]制備了具有磁性的復合新型三金屬氧化物Co3MnFeO6納米顆粒,作為非均相催化劑活化PMS降解有機污染物卡馬西平(CBZ). 吸附到催化劑表面的HSO5-被還原性的Co(Ⅱ)、Mn(Ⅱ)、Mn(Ⅲ)、Fe(Ⅱ)活化,通過電子轉移生成SO4·-. Co和Mn之間的氧化還原循環以及再生的Co2+和Mn2+會連續激活PMS,生成具有強氧化性的SO4·-,SO4·-與H2O或OH-之間相互作用生成·OH,與單金屬氧化物或雙金屬氧化物相比,三金屬氧化物的催化活性更高. Co、Mn和Fe在Co3MnFeO6催化劑中的協同作用是活化PMS高效生成SO4·-的主要原因. Co和Mn作為主要的活性位點,活化PMS產生自由基;Fe使Co3MnFeO6催化劑具有磁性,成為PMS和CBZ的吸附位點. 3種金屬之間的氧化還原循環使Co3MnFeO6催化反應高效持續進行,表現出較高的催化性和穩定性[73]. 綜上,錳基氧化物活化PMS的普遍機理與錳的氧化還原循環有關〔見圖1(b)〕.
4 錳基氧化物活化PDS的機制
PMS水溶液呈酸性且活性自由基的產生量相對較低,而PDS水溶液通常呈中性,易儲存且環境友好. 相較于活化PMS,除SO4·-和·OH在PDS體系中作為主要的反應物質外,還存在單獨生成1O2的氧化反應路徑.
4.1 MnO2活化PDS
與PMS反應機理相似,MnO2也被用于活化PDS降解硝基苯[74]和2,4-二氯苯酚[75]等污染物. SO4·-和·OH是α-MnO2活化PDS的主要活性物種. 然而,PDS/β-MnO2體系降解苯酚是一種非自由基的氧化反應[76]. 與β-MnO2活化PMS產生SO4·-、·OH和1O2不同,β-MnO2活化PDS僅產生1O2. Fang等[77-78]認為,1O2的產生可能是由于超氧離子和自由基的直接氧化或重組,MnO2的表面會形成一種中性的亞穩態錳中間體(MnⅣ—O—O—SO3),與S2O82-反應并伴隨MnⅣ—O斷裂形成超氧化物中間體O2·-[79],隨后MnⅣ直接氧化O2·-生成1O2[80](見圖2).
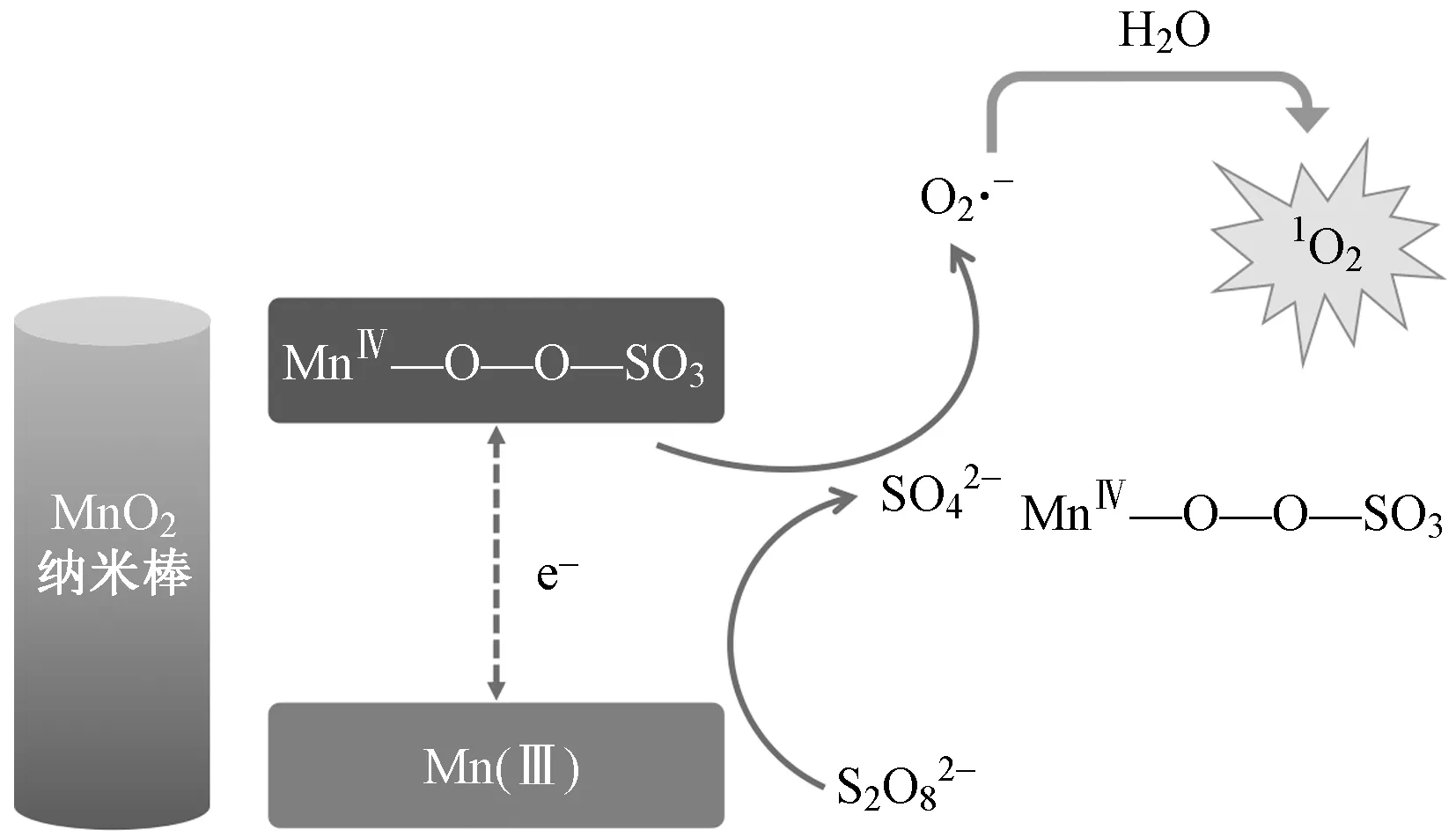
圖2 MnO2納米棒活化PDS生成單態氧的反應機理[80]Fig.2 The reaction mechanism of activation of PDS by MnO2 nanorods to generate singlet oxygen[80]
4.2 MnOx及其復合型氧化物活化PDS
除MnO2之外,MnOx(MnO、Mn3O4、Mn2O3和納米零價Mn等[81])也可以用于活化PDS降解污染物〔見式(25)(26)〕. MnOx與其他金屬氧化物復合能夠有效提高催化反應活性.
(25)
(26)
Shah等[82]利用Mn0、Mn(Ⅱ)、Mn(Ⅲ)活化S2O82-生成SO4·-和·OH降解環丙沙星(CIP). 由于生成了反應性較弱的S2O8·-以及部分電子從S2O82-轉移到金屬離子表面,使Mn(Ⅲ)/S2O82-降解CIP的效率較低. Mn0可再次生成Mn(Ⅱ),從而保證Mn(Ⅱ)持續充足的供給. 并且Mn0的表面積較大,能夠將目標污染物CIP吸附至表面從而提高催化活性. Li等[83]證明在3種Mn(Ⅲ)Ox中,γ-MnOOH相較于α-Mn2O3和Mn3O4在降解苯酚方面表現出更好的催化反應活性. 在中性和堿性條件下,SO4·-和·OH是降解苯酚的主要活性物質,而在酸性條件下苯酚的降解主要是γ-MnOOH和PDS反應形成的氧化中間體引起的. γ-Fe2O3/Mn3O4納米復合材料表面Fe(Ⅲ)被還原成Fe(Ⅱ)后與S2O82-發生類Fenton反應,生成SO4·-和Fe(Ⅲ)[84]. 隨著Mn3O4的引入,Mn(Ⅱ)充當電子轉移體,促進Fe(Ⅱ)的再生,提高反應活性[85].
5 結論與展望
a) 錳基氧化物的合成存在多種方法,在選擇有效的合成方法之前,要充分考慮錳基氧化物的表面結構、物理化學性質、濃度等因素. 通過改變反應時間、反應溫度、溶液中Mn的總濃度等,能夠調控錳氧化物的合成路徑. 水熱法是其中最常用方法之一,單獨使用水熱法合成錳基材料或與其他方法相結合,均可以制備具有較高催化反應活性的錳氧化物催化劑.
b) 水環境中存在的大量無機陰離子(HCO3-、NO3-、SO42-、CO32-等)與SO4·-或·OH反應,抑制水體中污染物的降解去除. 溶液的pH對反應性能的影響不顯著,反應溫度通常與有機污染物的降解率呈正相關,污染物的初始濃度與降解率呈負相關. 此外,晶型結構、表面積、氧化態也是影響錳基氧化物反應活性的重要因素.
c) 錳基氧化物活化PMS/PDS存在自由基(SO4·-和·OH)和非自由基(1O2)2種反應機理. 錳基氧化物活化PMS/PDS產生SO4·-,而·OH可由SO4·-與H2O或OH-反應生成. 同時,復合型錳基氧化物相較于單一MnO2具有更好的催化反應活性. MnO2活化PMS與PDS的反應機理不同:Mn(Ⅳ)被PMS還原生成SO5·-和Mn(Ⅲ),隨后被氧化成Mn(Ⅳ)并生成SO4·-;活化PDS是Mn(Ⅳ)與Mn(Ⅲ)之間的氧化還原,先生成S2O8·-后產生SO4·-. 此外,MnO2活化PDS存在僅產生1O2的非自由基反應機理.
d) 相比于活化PMS,利用錳基氧化物活化PDS降解廢水中有機污染物的研究較少,且二者的降解機制不同. 大多數錳基氧化物活化PMS過程通常會產生SO4·-和·OH,但β-MnO2活化PDS僅產生1O2. 未來需深入研究錳基氧化物活化PDS的機理.
e) 錳基氧化物經多次循環使用后仍具有較高的催化活性和穩定性. 但目前除了利用磁性對部分金屬氧化物進行回收外,大多使用過濾法. 因此,研發高效、經濟實用且易于快速分離的催化劑具有重要意義. 較多的復合型錳基氧化物催化劑已被證實具有較好的催化活性,但復合型氧化物催化劑的合成工藝較一元氧化物更為復雜. 在較溫和的條件下合成高效的復合氧化物催化劑可作為未來研究方向之一.
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現代企業(2015年9期)2015-02-28 18:56:50
