基于UPLC-Q-TOF-MS技術的當藥化學成分分析
2021-09-06 03:44:44段吉平段瓊2a王鐵戰馬春艷劉永利馮麗
中國現代中藥 2021年7期
關鍵詞:分析
段吉平,段瓊,2a,王鐵戰,馬春艷,劉永利*,馮麗
1.河北省藥品檢驗研究院,河北 石家莊 050011;
2.河北醫科大學,河北 石家莊 050017
當藥Swertiae Herba 為龍膽科獐牙菜屬瘤毛獐牙菜Swertia pseudochinensisHara 的干燥全草[1],于夏、秋季節采挖,在河北、內蒙古藥用時曾被稱為“肝炎草”。《中華人民共和國藥典》2020 年版中記載其味苦,性寒,歸肝、胃、大腸經,用于濕熱黃疸、脅痛、痢疾腹痛、食欲不振[2]。其在《內蒙古中草藥》[3]、《中華本草》[4]、《全國中草藥匯編》[5]、《河北植物志》(第二卷)[6]中均有記載。目前,對當藥的研究相對較少,化學成分方面也鮮見報道。本實驗采用超高效液相色譜-四級桿-飛行時間質譜法(UPLC-Q-TOF-MS)對當藥進行化學成分分析,同時對多類別的成分進行結構鑒定及確認,為當藥化學成分和藥效物質基礎研究及多元質量評價方法的建立提供參考。
1 材料
1.1 試藥
當藥藥材購自安國藥材市場,經河北省藥品檢驗研究院牛小蓮主任藥師鑒定為龍膽科獐牙菜屬瘤毛獐牙菜Swertia pseudochinensisHara的干燥全草。
對照品龍膽苦苷(批號:770-200105)、當藥苷(批號:111742-200501)、獐牙菜苦苷(批號:0785-200203)、芒果苷(批號:111607-201503)、齊墩果酸(批號:110709-201808)、甜菜堿(批號:110894-200503)和槲皮素(批號:100081-201610)均購自中國食品藥品檢定研究院,純度均大于99%。
甲醇(分析純,天津科密歐化學試劑有限公司);質譜用甲酸(Fisher 公司);乙腈和甲醇(色譜純,Merck公司)。
1.2 儀器
LC-TripleTOF?5600+型液質聯用儀(SCIEX 公司),配備Analyst?TF 1.6 質譜操作軟件、PeakView 2.0 質譜圖分析軟件;AG245 型電子天平(Mettler Toledo公司);KQ-400KDE型超聲清洗儀(昆山超聲儀器有限公司)。
2 方法
2.1 色譜條件
色譜柱為菲羅門Kinetex XB-C18100A(100 mm×2.1 mm,2.6 μm)。流動相為0.1% 甲酸水溶液(A)-乙腈(B),梯度洗脫(0~4 min,15%B;4~7 min,15%~30%B;7~10 min,30%~40%B;10~11 min,40%~50%B;11~30 min,50%~70%B;30~40 min,70%~100%B;40~42 min,100%B;42.1~45 min,15%B)。柱溫:20 ℃,流速:0.3 mL·min-1,進樣量:1 μL。
2.2 質譜條件
采用電噴霧離子源(ESI),正離子和負離子掃描模式。正離子模式下電噴霧電壓為5500 V,氣簾氣(CUR)為30 psi(1 psi≈6.79 kPa,下同),去簇電壓(DP)為80 V,碰撞能量(CE)為10 V;負離子模式下電噴霧電壓為4500 V,CUR 為30 psi,DP為80 V,CE為10 V。掃描質量數為m/z100~1000。
2.3 溶液的制備
2.3.1 對照品溶液制備 取7 種對照品適量,分別加甲醇制成質量濃度為10 μg·mL-1的單一對照品溶液。
2.3.2 供試品溶液制備 取當藥適量,粉碎,過三號篩,混勻,取0.5 g,精密稱定,置具塞錐形瓶中,精密加入甲醇25 mL,密塞,稱定質量,超聲處理15 min,放冷,再稱定質量,用甲醇補足減失質量,搖勻,用0.22 μm微孔濾膜濾過,即得。
2.4 化學成分分析
按2.1 和2.2 項下分析條件,對當藥樣品進行正、負離子模式分析,使用PeakView 2.0 質譜圖分析軟件對所得到的樣品進行處理并對色譜峰進行同位素及MS/MS 碎片分析。首先,與對照品圖譜中各已知成分的相對保留時間(tR)、準分子離子、二級質譜碎片等進行比對,再結合現有文獻中同屬植物的化學物質建立樣品的Session 篩選庫[7],對樣品中的色譜峰進行解析,以滿足質量誤差<5×10-6、同位素分布正確且有二級碎片的化合物作為目標物質。逐一分析物質的同位素信息和MS/MS碎片碎裂規律,并結合Formula Finder 分析結果,推斷可能的候選化合物[8-15]。
3 結果
3.1 色譜峰的鑒定
當藥樣品正、負離子模式下得到的總離子流圖(TIC)見圖1。共確認29 個化合物,包括7 個環烯醚萜類、13 個酮類、4 個黃酮類、2 個三萜類、2 個生物堿類和1 個核苷類成分。其中10 個為當藥中未見報道的成分,見表1。

表1 當藥中確認的29個成分信息
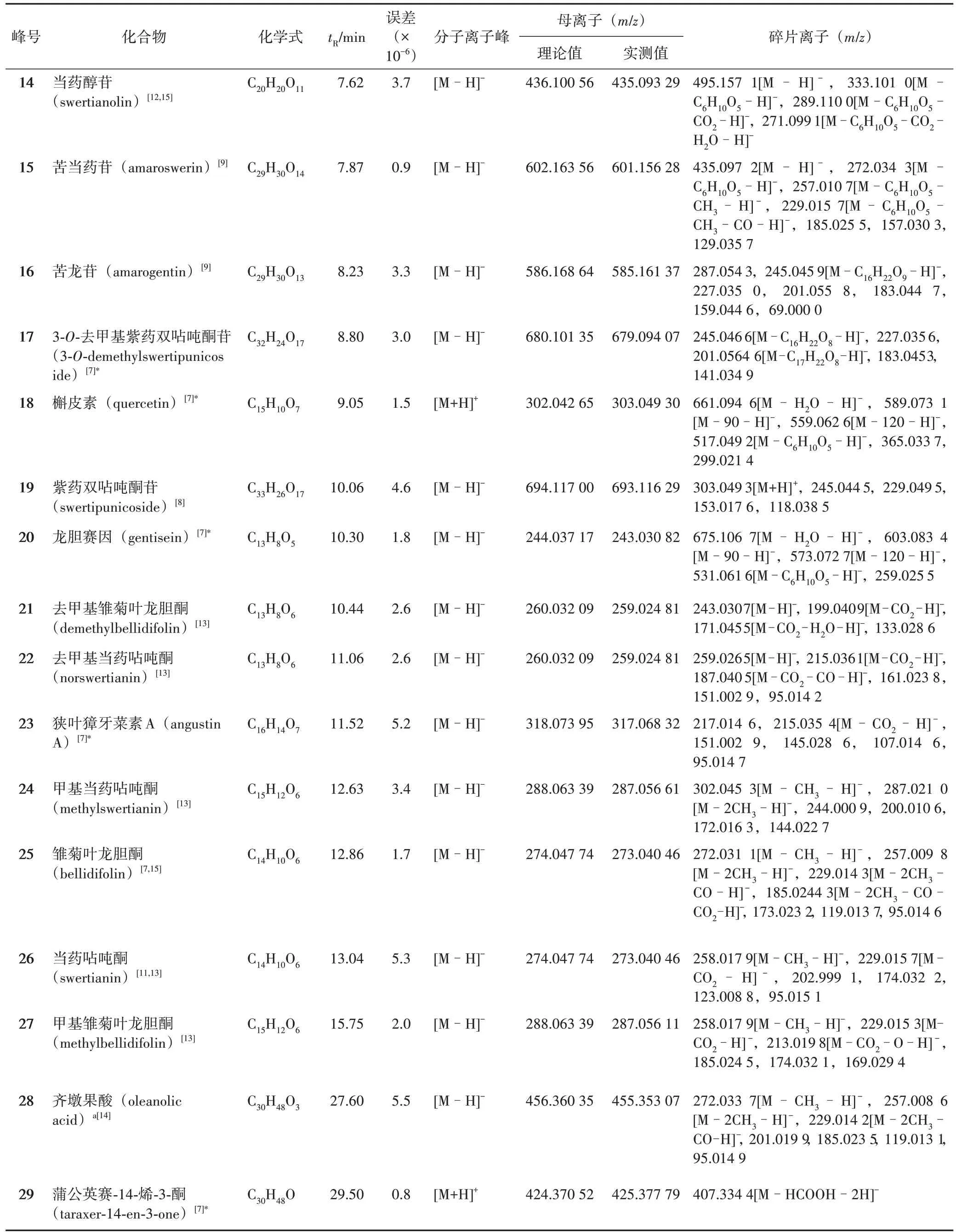
續表1
3.2 各類化合物分析鑒定
3.2.1 環烯醚萜類化合物 從當藥中鑒定出的環烯醚萜類成分主要有獐牙菜苦苷、龍膽苦苷、當藥苷、苦當藥苷、苦龍苷、馬錢苷酸和日本獐牙菜苷7 個成分。環烯醚萜類成分主要具有特征的二氫吡喃環順式連接1 個五元環的單元結構,而當藥中主要存在環戊烷型和裂環烯醚萜苷2 種類型。在所采用的色譜系統中加入了甲酸,故環烯醚萜類物質準分子離子峰存在[M+HCOO]-及[M-H]-2 種形式。當藥中的環烯醚萜類結構均存在-C6H10O5碎片丟失,同時因裂環烯醚萜苷類化合物具有半縮醛結構,化學性質活潑,在糖苷鍵斷裂后繼而發生逆狄爾斯-阿德爾(RDA)裂解,失去-CO2、-H2O 等碎片,同時可能發生-CH3的丟失。以當藥苷為例,m/z403.123 5[M+HCOO]-與m/z357.119 2[M-H]-同時存在于系統中,但[M+HCOO]-響應明顯強于[M-H]-。在丟失-C6H10O5碎片后,繼而發生RDA 裂解,脫去-CO2,得到m/z151.075 8[M-C6H10O5-CO2-H]-。
3 號色譜峰保留時間為3.70 min,根據PeakView 2.0 質譜圖軟件分析化學式為C16H24O10,理論質量數為376.136 95[M],在負離子檢測模式下實際測得質量數為m/z375.131 63[M-H]-。其進一步脫葡萄糖基產生m/z213.076 1[M-C6H10O5-H]-,由于羧基的不穩定性,[M-C6H10O5-H]-脫去羧基得到m/z169.087 0[M-C6H10O5-CO2-H]-,再脫去中性小分子H2O,得到m/z151.075 3 [C9H11O2]-。該化合物與文獻報道的馬錢苷酸碎片信息一致[17],推測其為馬錢苷酸。從其MS/MS 圖中所得到的碎片離子信息可解析其裂解方式,見圖2。
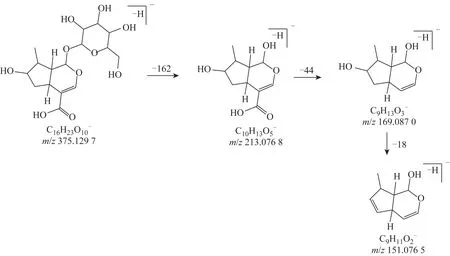
圖2 馬錢苷酸裂解途徑

圖3 當藥中的11個單酮及酮苷

圖4 當藥呫噸酮裂解途徑

圖5 3-O-去甲基紫藥雙呫噸酮苷裂解途徑
具有O苷鍵的酮苷中,因糖苷鍵的不穩定性,首先生成帶1個負電荷的酮苷元。12號色譜峰保留時間為7.00 min,根據PeakView 2.0質譜圖軟件分析化學式為C26H30O16,理論質量數為598.153 39[M],在負離子檢測模式下實際測得為m/z597.146 11[M-H]-。母離子脫去-C12H20O10,生成的碎片離子為m/z273.041 1[M-C12H20O10-H]-。m/z273.041 1[M-C12H20O10-H]-在丟失-CH3碎片后,繼而丟失-CO,產生m/z258.017 7[M-C12H20O10-CH3-H]-及m/z230.022 7 [MC12H20O10-CH3-CO-H]-的碎片。推測該化合物可能為紫藥雙呫噸酮苷。
3.2.3 黃酮類化合物 當藥中主要鑒定出4 個黃酮類化合物,包括當藥黃素、異牡荊素、異葒草素和槲皮素。在負離子掃描條件下,黃酮類成分易中性丟失-CO、-CO2,并可能在糖基的部分發生RDA裂解。10號色譜峰保留時間為6.16 min,根據PeakView 2.0質譜圖軟件分析化學式為C22H22O11,理論質量數為462.116 21[M],在負離子檢測模式下實際測得質量數為m/z461.108 94[M-H]-。母離子發生RDA反應,生成了m/z341.068 8[M-120-H]-的碎片,隨后又丟失-CO及-CH3,得到m/z313.037 2[M-120-CO-H]-和m/z298.050 2[M-120-CO-CH3-H]-。推測該化合物為當藥黃素。
3.2.4 三萜類化合物 28 號色譜峰保留時間為27.60 min,根據PeakView 2.0 質譜圖軟件分析化學式為C30H48O3,其一級和二級質譜圖與齊墩果酸對照品一致。齊墩果酸理論質量數為m/z456.360 3[M],在負離子檢測模式下實際測得質量數為m/z455.353 07[M-H]-,母離子經過脫羧反應、失去活潑氫和1 分子H2O,形成了共軛結構的穩定碎片離子m/z407.334 4[M-H-HCOOH-2H]-和m/z391.304 3[MHCOOH-H2O-H]-碎片離子。
29 號色譜峰保留時間為29.50 min,根據PeakView 2.0 質譜圖軟件分析化學式為C30H48O,理論質量數為424.370 52[M]。在正離子檢測模式下測得質量數為m/z425.377 79[M+H]+。母離子發生RDA裂解,生成m/z301.258 3[M-124]+的碎片,推測其結構為蒲公英賽-14-烯-3-酮。
3.2.5 生物堿類化合物 1 號色譜峰保留時間為0.74 min,根據PeakView 2.0質譜圖軟件分析化學式為C5H11NO2,其保留時間、一級和二級質譜圖與甜菜堿對照品一致。甜菜堿理論質量數為m/z117.078 98[M],在正離子模式下,實際測得質量數為m/z118.086 26[M+H]+,母離子發生化學鍵斷裂及重排失去m/z59.073 5[N(CH3)3]+和m/z60.021 1 [CH3COOH]+的碎片,得到m/z59.076 2[M-N(CH3)3+H]+和m/z58.070 4[M-CH3COOH+H]+的碎片離子。
8 號色譜峰保留時間為5.30 min,根據PeakView 2.0質譜圖軟件分析化學式為C10H9NO2,理論質量數為175.063 33[M],在正離子模式下,實際測得質量數為m/z176.070 61[M+H]+。母離子發生化學鍵斷裂及重排失去HCHO,得到m/z146.060 0[MHCHO+H]+,隨后[M-HCHO+H]+丟失CO,得到m/z118.066 2 的碎片離子,推測結構為龍膽堿,裂解途徑見圖6。

圖6 龍膽堿的裂解途徑
3.2.6 核苷類化合物 29 號色譜峰保留時間為1.22 min,根據PeakView 2.0 質譜圖軟件分析化學式為C10H13N5O4,理論質量數為267.096 75[M],在正離子模式下,實際測得質量數為m/z268.104 41[M+H]+,母離子發生化學鍵斷裂及重排失去C5H9O4,得到m/z136.062 4的碎片離子。推測結構為腺苷。
4 討論
本研究應用UPLC-Q-TOF-MS、Analyst?TF 1.6 Software 質譜操作軟件和PeakView 2.0 質譜圖分析軟件對當藥的化學成分進行分析,建立了獐牙菜屬植物的物質篩選庫,對樣品中色譜峰進行解析,分析物質的同位素信息和MS/MS 碎片信息,與對照品圖譜中各已知成分的保留時間、準分子離子、二級質譜碎片等進行比對,再結合現有文獻中同屬植物的化學物質[7],對樣品中的色譜峰進行解析。最終確認了當藥中的7個環烯醚萜類、13個酮類、4個黃酮類、2 個三萜類、2 個生物堿類和1 個核苷類成分。其中馬錢苷酸、龍膽堿、紫藥雙呫噸酮苷、日本獐牙菜苷、3-O-去甲基紫藥雙呫噸酮苷、槲皮素、龍膽賽因、狹葉獐牙菜素A、腺苷和蒲公英賽-14-烯-3-酮為首次在當藥中報道。
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
