基于高通量測序分析牛呼吸系統疾病鼻咽微生物多樣性和功能預測研究
2021-09-06 05:44:50張靜張濤盧昱希任榮清黃波周思旋王婧趙春萍史開志張雄
畜牧與獸醫 2021年9期
關鍵詞:功能
張靜,張濤,盧昱希,任榮清,黃波,周思旋,王婧,趙春萍,史開志,張雄*
(1.貴州省農業科學院畜牧獸醫研究所/畜禽重大疫病監測防治重點實驗室,貴州 貴陽 550002;2.貴州省農業科學院畜牧獸醫研究所/獸用中草藥研究室,貴州 貴陽 550002)
牛鼻咽部是連接外部環境與機體內環境的重要部位,也是牛呼吸系統疾病(bovine respiratory disease, BRD)致病微生物入侵機體的切入點。據調查,我國養牛業中65%的疾病為BRD,其中肉牛場的感染率達90%以上,死亡率達35%[1]。而這些被診斷患有BRD的牛主要以抗生素治療為主,大量抗生素的使用產生耐藥菌株,最終導致無藥可治。因此,系統性地了解患BRD牛鼻咽部微生物群落特征及致病性病原體有助于定向治療呼吸系統疾病,減少抗菌藥物濫用。然而,牛鼻咽部是一個半開放的環境,過境微生物種類繁多,且存在許多不可離體培養的條件性致病微生物,利用傳統的培養技術和常規PCR檢測來探索牛上呼吸道微生物多樣性、分析其功能具有一定局限性;細菌16S rRNA、真菌18S rRNA和真菌核糖體基因轉錄間隔區(internal transcribed spacer,ITS)測序技術的發展使基于培養方法難以或不可能檢測到的微生物物種的鑒定和定量成為可能[2-4]。
近年來,國內外利用微生物擴增子測序對牛消化系統微生物組成和性質,以及微生物群落結構的變化如何影響宿主的健康進行了廣泛研究[2,5]。與消化系統微生物群在胃腸道中的作用相似,呼吸道微生物群也是影響呼吸系統健康的重要因素[6]。健康的牛呼吸道菌群處于動態平衡,主要由變形桿菌門(Proteobacteria)、厚壁菌門(Firmicutes)、軟壁菌門(Tenericutes)、放線菌門(Actinobacteria)和擬桿菌門(Bacteroidetes)5個門水平下的500余種細菌種屬組成,每種細菌種屬的相對含量較低[7]。牛鼻咽微生物多樣性及每類細菌的相對豐度隨牛群所處的生產系統、年齡及健康狀況的變化而變化[8]。Holman等[6]采用454焦磷酸測序技術評估了引種第1天和60 d后健康的牛上呼吸道微生物多樣性,剛進入飼養場的牛群鼻咽微生物群主要由假單胞菌屬(Pseudomonas)、希瓦氏菌屬(Shewanella)、不動桿菌屬(Acinetobacter)和肉桿菌屬(Carnobacterium)等構成,易發生呼吸系統疾病;飼養60 d后牛鼻咽部微生物則逐漸趨于一致,主要以葡萄球菌屬(Staphylococcus)、分支桿菌屬(Mycobacterium)、曼氏桿菌屬(Mannheimia)和莫拉菌屬(Moraxella)為主,2個時間點牛群鼻咽微生物群之間存在顯著差異。研究還發現,乳桿菌(Lactobacillus)/乳球菌(Lactococcus)豐度增加表明宿主處于健康狀態,而溶血性曼氏桿菌(M.haemolytica)、支原體(Mycoplasmabovis)及多殺性巴氏桿菌(Pasteurellamultocida)等相對豐度增加則表示宿主處于疾病狀態[9-10]。Zeineldin等[11]也發現,門水平下,BRD牛鼻咽部變形桿菌門(32.12%)、放線菌門(38.2%)和梭桿菌門(3.86%)比健康牛多;屬水平下,BRD牛不動桿菌屬(Acinetobacter,12.54%)、梭菌屬(Fusobacterium,3.71%)和巴氏桿菌屬(Pasteurella,2.38%)含量比健康牛高,支原體和莫拉菌屬的含量顯著高于健康牛。在BRD致死的牛肺灌洗液和肺組織中,致病性病原菌,如溶血性曼氏桿菌(M.haemolytica)、支原體及昏睡嗜血桿菌(H.somnus)的相對豐度都大于5%,其他種屬在1%~5%之間,而健康的牛肺灌洗液中未發現支原體[3,12]。由此可知,鼻咽微生物的組成及豐度能直接影響牛下呼吸道中的微生態平衡,導致牛呼吸道疾病加劇。部分研究還表明真菌在慢性呼吸道疾病的發生和發展過程中也具有重要作用,且易產生復雜的耐藥性[13]。張傳師等[14]總結了國外的相關研究主要集中在鼻咽部細菌類微生物組與BRD的關系上,缺乏對牛呼吸道真菌微生物組學的研究,在國內則未見對患呼吸系統疾病牛鼻咽部微生物群的相關研究報道。因此,從鼻咽部細菌組和真菌組群落組成出發,全面了解并闡述微生物組與BRD之間的關系,有助于進一步探討疾病發病機制,輔助臨床防治工作。
本研究以2019—2020年期間采集的貴州省內3個牛場患BRD的牛鼻咽微生物為研究對象,利用16S rRNA V3-V4區和ITS1區Illumina NovaSeq測序,橫向比較不同地區患病牛鼻咽部微生物群落組成并預測其功能,以期為系統了解養牛場發病病因,深入研究鼻咽微生物的組成與宿主呼吸系統健康之間的關系,開發新型微生態制劑及指導牛呼吸系統疾病防制工作提供基礎數據。
1 材料與方法
1.1 樣品采集
R1組共14份樣品,2020年4月采自貴州省盤縣某牛場,該牛場患病牛臨床表現為中等以上呼吸系統疾病癥狀,具體表現為咳嗽、鼻腔分泌物多,嚴重時鼻腔有出血現象,持續時間10~20 d;R2組共6份樣品,2020年5月采自貴州省盤縣某牛場,該牛場患病牛臨床表現為咳嗽,鼻腔分泌物增加,食欲減退,精神不佳的癥狀,持續時間為5~15 d;R3組共12份樣品,2019年12月采自貴州省大方縣某牛場,該牛場患病牛臨床表現為流鼻涕、輕微咳嗽、食欲減退,持續時間為10~15 d。用無菌棉簽采集患病牛鼻腔拭子,于無菌管中保存,放置液氮罐中送回實驗室于-80 ℃保存備用。
1.2 樣品中DNA的提取及PCR擴增
利用十六烷基三甲基溴化銨(CTAB)法對鼻拭子樣品基因組DNA進行提取。之后利用瓊脂糖凝膠電泳檢測DNA的純度和濃度,取適量的樣本DNA于離心管中,使用無菌水稀釋樣本至1 ng/μL。以稀釋后的基因組DNA為模板,擴增細菌16S V3-V4高變區(341F:5′-CCTAYGGGRBGCASCAG-3′, 806R:5′-GGACTACNNGGGTATCTAAT-3′)和真菌ITS1區(ITS1-1F-F:5′-CTTGGTCATTTAGAGGAAGTAA-3′,ITS1-1F-R:5′-GCTGCGTTCTTCATCGATGC-3′),文庫構建及Ilumina NovaSeq測序由北京諾禾致源科技股份有限公司協助完成。
1.3 測序數據處理
將每個樣本測序獲得的讀段(reads)進行拼接,得到的拼接序列為原始標簽(raw tags)數據。將原始標簽序列經過嚴格的過濾處理得到高質量的標簽數據(clean tags)。利用QIIME[15]軟件對高質量的標簽數據進行質量控制,具體操作如下:①標簽截取:將原始標簽從連續3個以上堿基質量小于或等于19的第1個低質量堿基位點截斷;②標簽長度過濾:標簽數據經過截取后得到的標簽數據集,進一步過濾掉其中連續高質量堿基長度小于標簽長度75%的標簽序列。質控后的標簽序列去除其中的嵌合體序列,得到最終的有效數據(effective tags)進行數據分析。
1.4 生物信息學分析
利用Uparse[16]對所有樣本的有效序列以97%的一致性聚類成為分類操作單元(operational taxonomic units,OTUs),為檢驗測序數據是否達到飽和且符合后續分析要求,從樣本中隨機抽取一定測序量的數據,統計它們所代表OTUs數目,以抽取的測序數據量與對應的物種數來構建稀釋曲線(rarefraction curve),并根據樣本中OTUs的相對豐度繪制等級聚類曲線(rank abundance curve)。使用QIIME軟件進行樣本復雜度分析(alpha diversity),包括Chao1,ACE,Shannon,Simpson和Observed species共5個指數;計算Unifrac距離,進行多組間比較分析(beta diversity)并繪制PCoA圖。對OTUs序列進行物種注釋,用Mothur[17]和QIIME分別對細菌和真菌進行物種注釋,在門和屬分類水平上,統計不同組中的物種豐度和多樣性,為進一步挖掘組間微生物菌落的差異,選用t檢驗對3組樣本兩兩之間的微生物菌落組成進行統計分析,篩選出各組存在顯著差異的菌落。
1.5 16S rRNA和ITS功能預測分析
為探討牛鼻咽部微生物群落在其體內發揮的功能,使用PICRUSt[18]和FunGuild[19]方法分別對細菌和真菌進行了功能預測,利用R軟件繪制每組樣本的豐度信息熱圖,并從功能差異層面進行聚類。
1.6 數據的統計與分析
Alpha多樣性指數組間差異分析選用Tukey檢驗,結果表示為“平均數±標準差”,P<0.05表示差異顯著,P<0.01表示差異極顯著。
2 結果與分析
2.1 序列拼接質控與OTUs聚類分析
經16S rRNA和ITS基因高通量測序,平均測得牛上呼吸道內細菌原始序列為87 696條,經質控后平均得到64 128條有效序列;R1、R2和R3組對應的平均有效序列分別為64 880、63 545和63 959條。平均獲得真菌原始序列105 973條,經質控后,平均得到有效序列65 472條;R1、R2和R3組對應的平均有效序列分別為64 285、65 560和66 572條(表1)。在97%相似度標準下聚類分析,分別得到細菌OTU為2 851、2 358和2 728個,其中3組樣本共有的OTU數目為1 683,R1、R2和R3組特有的OTU數目分別為474、225和353;得到的真菌OTU分別為1 766、1 213和2 125個,其中3組樣本共有的OTU數目為825個,R1、R2和R3組特有的OTU數目分別為263、139和607個(圖1)。

表1 牛鼻咽部細菌16S rRNA基因和真菌ITS高通量測序基本信息表

圖1 3組牛鼻咽部細菌16S rRNA(A)和真菌ITS(B)聚類OTUs韋恩圖
2.2 微生物群落的Alpha多樣性分析
16S rRNA和ITS測序數據的稀釋曲線和等級聚類曲線結果表明,各組樣本量足夠,測序深度已經基本覆蓋到樣品中所有物種(圖2、圖3)。Alpha多樣性指數反映樣品微生物群落的多樣性。其中,Chao1、ACE 指數反映菌群豐度,由表2可知,R1、R2和R3組之間上呼吸道細菌及真菌菌群豐度相似,R2組細菌和真菌的菌群豐度都最高,細菌菌群豐度最低的是R3組,真菌菌群豐度最低的是R1組。菌群的多樣性由Simpson和Shannon指數反映,數據顯示,細菌群落多樣性最高的是R2組,R1組次之,R3組最低;R1和R2組菌群多樣性顯著(P<0.05)高于R3組,R1組與R2組之間無顯著差異;真菌菌落多樣性最高的是R3組,其次是R2組,最低的是R1組;R1組菌群多樣性顯著(P<0.05)低于R3組,R1組與R2組之間無顯著差異。表明3組樣品中菌群豐度雖然相似,但菌落多樣性存在較大差異。

圖2 不同組別中的16S rRNA稀釋曲線(A)和等級聚類曲線(B)
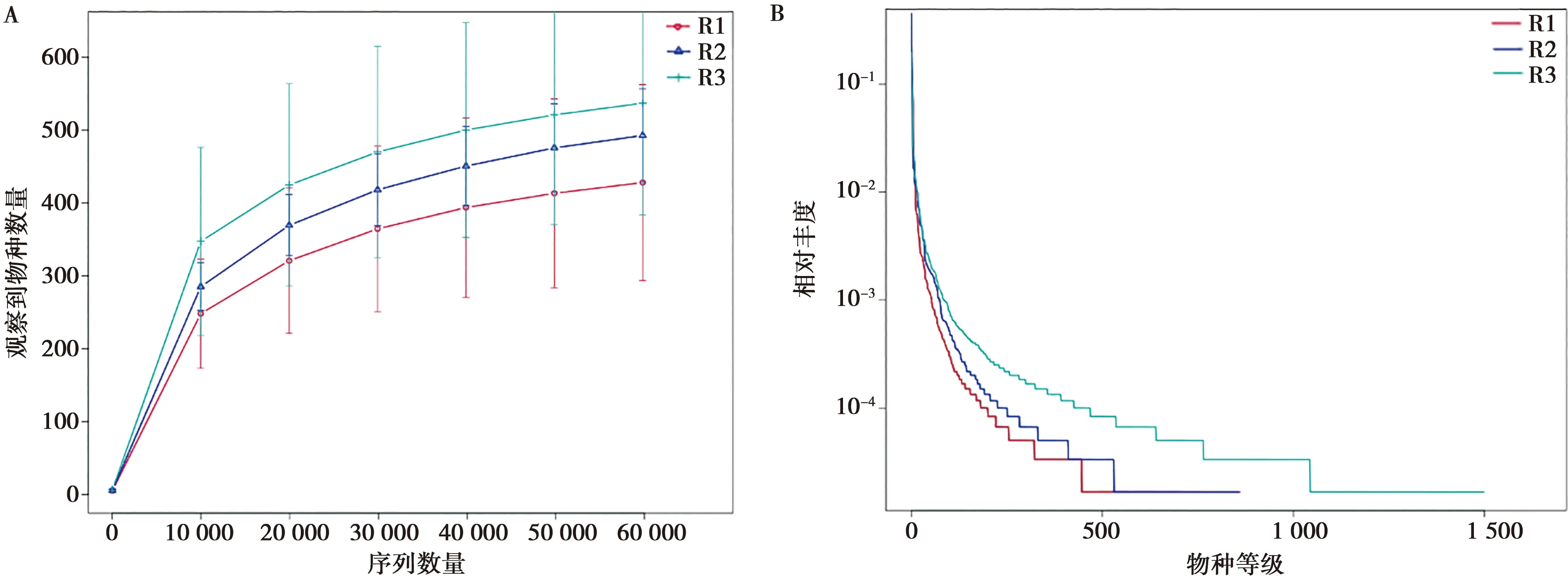
圖3 不同組別中的ITS稀釋曲線(A)和等級聚類曲線(B)

表2 3組樣本的Alpha多樣性指數
2.3 微生物群落的Beta多樣性分析
基于加權的Unifrac距離進行PCoA分析,分析3組樣品間微生物的相似性。由圖4A可知,細菌類主坐標PC1和PC2的貢獻率分別為總變量的38.43%和17.81%,3組樣品并沒有各自單獨聚類,說明9組樣品細菌微生物群落組成相似。而真菌類微生物PCoA分析結果顯示(圖4B),主坐標PC1和PC2的貢獻率分別為總變量的45.69%和23.87%,R1和R2組聚為一類,R3組則單獨聚類,表明R3組中的多數患病牛上呼吸道真菌群落組成與R1、R2組差異較大。
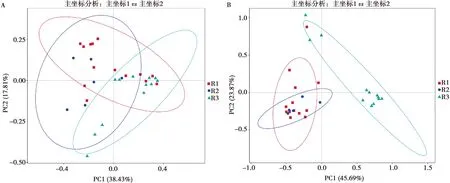
圖4 不同地區患病牛上呼吸道中細菌類微生物群落PCoA圖(A)和真菌類微生物群落PCoA圖(B)
2.4 不同地區患病牛上呼吸道中的微生物品種鑒定及菌群結構分析
3個不同牛場患病牛上呼吸道中細菌類微生物門水平分布如表3所示,主要以變形桿菌門(Proteobacteria)、擬桿菌門(Bacteroidetes)、梭桿菌門(Fusobacteria)、厚壁菌門(Firmicutes)、軟壁菌門(Tenericutes)及放線菌門(Actinobacteria)為主;與R1組相比,R2組變形桿菌門顯著(P<0.05)減少,R3組擬桿菌門、厚壁菌門及放線菌門顯著減少;與R2組相比,R3組變形桿菌門顯著增加,擬桿菌門則顯著減少。屬水平結果顯示,3組樣品中排前10的主要菌群屬組成相似,但相對豐度相差較大。R1組中主要以擬桿菌屬(Bacteroides)、不動桿菌屬、卟啉菌屬(Porphyromonas)及梭菌屬(Fusobacterium),其中不動桿菌屬和擬桿菌屬顯著高于R3組;R2組中主要以卟啉菌屬、擬桿菌屬、梭菌屬及不動桿菌屬,其中不動桿菌屬顯著高于R3組,R3組中主要以莫拉菌屬(Moraxella)、支原體屬(Mycoplasma)、梭菌屬及卟啉菌屬,其中莫拉菌屬顯著高于R1和R2組。
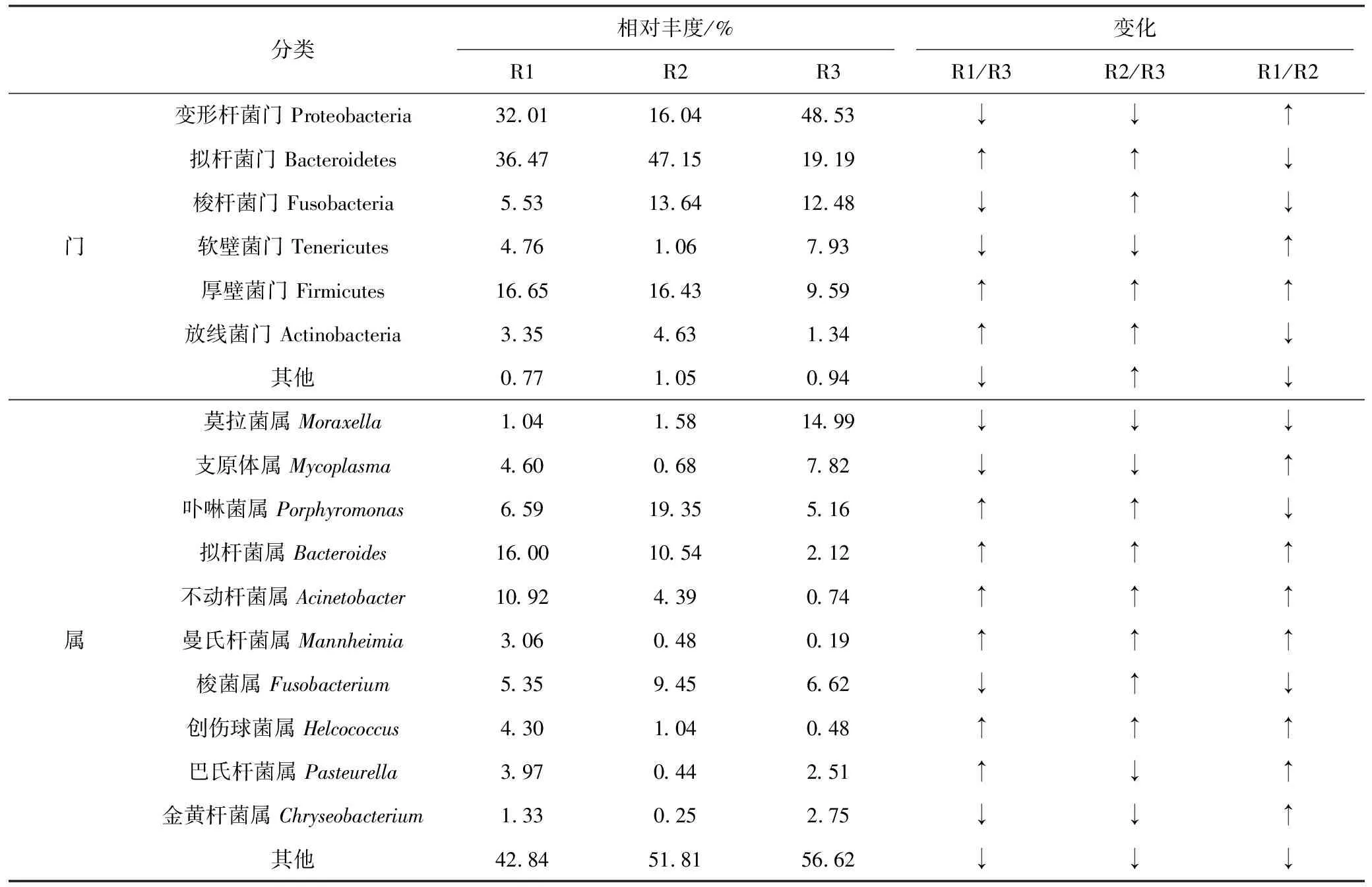
表3 細菌分類門水平和排前10的屬水平的相對分布
真菌類微生物門水平分布如表4所示,3組樣品中,主要以子囊菌門(Ascomycota)和擔子菌門(Basidiomycota)為主,R1組和R2組內的擔子菌門極顯著高于(P<0.01)R3組。屬水平上,3組樣品排前10的主要菌群屬豐度相差較大。R1組主要以克柔氏菌屬(Issatchenkia)、隱毛孢耳屬(Apiotrichum)、假絲酵母屬(Diutina)、毛孢子菌屬(Trichosporon)和曲霉菌屬(Aspergillus)為主;R2組主要以克柔氏菌屬、隱毛孢耳屬和毛孢子菌屬為主;R3組主要以曲霉菌屬、(Millerozyma)和克柔菌屬為主。與R1組相比,R2組假絲酵母屬相對豐度極顯著減少;與R3組相比,R1組和R2組克柔氏菌屬豐度增加,曲霉菌屬豐度減少,差異極顯著。

表4 真菌分類門水平和排前10的屬水平的相對分布
2.5 上呼吸道微生物功能預測
用PICRUSt對不同地區患病牛上呼吸道細菌類群進行功能預測。由圖5可知,KEGG通路二級功能層面上豐度排名前35的預測功能基因及它們在每個組中的豐度信息。R1組樣品有18個功能基因通路豐度減少,17個功能基因通路豐度增加;R2組樣品有12個功能基因通路豐度減少,23個功能基因通路豐度增加;R3組樣品有21個功能基因豐度減少,14個功能基因豐度增加。R1組和R2組功能基因豐度富集相似;然而,R3組中功能基因豐度增加的通路在R1和R2組中的豐度降低。其中,R3組中豐度增加的功能基因通路有:疾病感染、遺傳信息呈遞、轉錄、細胞過程和信號、信號轉導、癌癥和神經退行性疾病等相關通路;豐度減少的功能基因通路有:核苷酸代謝、細胞生長和死亡、消化系統、代謝疾病、氨基酸代謝和碳水化合物代謝等相關通路。結果表明,R1組和R2組患病牛感染的病原能夠導致代謝類的相關通路豐度增加,其中R2組豐度增加的通路比R1組多;疾病感染、信號轉導等相關通路豐度減少。而R3組患病牛感染的病原功能基因豐度富集的結果與R2組相反。

圖5 不同地區患病牛上呼吸道中細菌類微生物功能注釋聚類熱圖
基于真菌的物種分類,FunGuild可以獲得相應真菌的生態功能。用FunGuild對不同地區患病牛上呼吸道真菌類群進行功能預測。由圖6可知,3組樣品的上呼吸道中均存在動物病原菌富集及豐度增加;其中R1和R2組中的動物病原菌分別有2種,R1組種有1種,3組樣品中的病原菌種類各不相同。
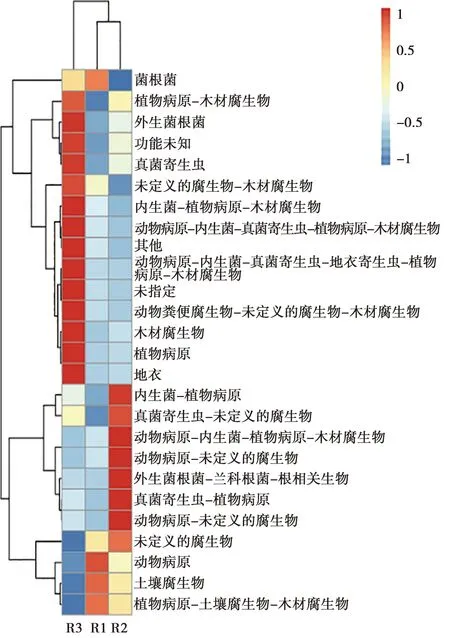
圖6 不同地區患病牛上呼吸道中真菌類微生物功能注釋聚類熱圖
3 討論
3.1 呼吸系統疾病牛鼻咽部菌群多樣性
本研究通過高通量測序技術對患BRD的牛鼻咽部菌群結構及組成進行分析,并基于細菌和真菌注釋數據庫進行菌群功能預測。不同場中微生物Alpha多樣性分析和組間微生物群落Beta多樣性分析結果表明,不同發病牛場細菌和真菌豐度相似;同一地區不同牛場菌群多樣性及菌落結構組成相似,但不同地區牛場之間菌群多樣性存在顯著差異,菌落結構組成也有所不同。由此可知,牛慢性呼吸系統疾病受地區環境因素影響較大,不同地區導致發病的誘因存在差異。臨床上,需進一步采用分子生物學技術或微生物菌屬注釋確認病原后進行針對性的治療,否則會導致牛場抗生素濫用,產生耐藥性。除此之外,隨著生物安全控制體系在畜禽疫病防控上獲得的顯著成效,牛場生物安全控制體系的建立也應該被重視,使其成為控制和預防地區性牛呼吸系統疾病蔓延的關鍵。
本研究結果顯示,患病牛呼吸道細菌類微生物主要由6個門類組成,與Zeineldin等[11]研究的BRD犢牛鼻咽優勢菌群(變形桿菌門32.12%、放線菌門38.2%和梭桿菌門3.86%)組成相比,本研究的樣品中還存在擬桿菌門(19.19%~47.15%)、厚壁菌門(9.59%~16.65%)和軟壁菌門(1.06%~7.93%);而放線菌門相對豐度較低,梭桿菌門相對豐度較高,可能是由不同地區及不同環境所致。與正常肉牛鼻咽部微生物組成[20]相比,患BRD牛能檢測到擬桿菌門和梭桿菌門的存在,放線桿菌門和軟壁菌門的相對豐度增加,厚壁菌門和變形桿菌門相對豐度類似,說明BRD可導致部分鼻咽部菌群組成及相對豐度變化。屬水平上,患BRD牛鼻咽部微生物相對豐度均小于20%,其中包括莫拉菌屬(1.04%~14.99%)、卟啉菌屬(5.16%~19.35%)、擬桿菌屬(2.12%~16%)、不動桿菌屬(0.74%~10.92%)、支原體屬(0.68%~7.82%)和曼氏桿菌屬(0.19%~3.06%)。與大多數研究結果[8]一致,支原體屬、莫拉菌屬、曼氏桿菌屬、巴氏桿菌屬在不同地區的患BRD牛鼻咽部中均存在,推測這3種菌屬可能在牛呼吸系統疾病中起到關鍵作用,多重感染的具體作用機制仍需進一步研究。
研究表明,真菌在人類呼吸系統疾病的發生、發展過程中扮演著重要角色,如真菌與喘息病、耐藥性等相關,也參與了宿主黏膜免疫反應,并證實了呼吸系統疾病的發生會導致真菌群的變化[21]。本研究對患BRD牛鼻咽部真菌類微生物進行了探究,發現與慢性呼吸系統疾病相關的優勢菌屬,如克柔氏菌屬(2.28%~45.1%)、曲霉菌屬(1.19%~16.26%)等,表明患BRD的牛呼吸系統會受到致病性真菌的入侵,最終導致真菌性感染。
3.2 呼吸系統疾病牛鼻咽部菌群中的致病菌
Lima等[9]利用高通量測序技術對健康和患BRD牛的鼻咽微生物進行了分析,與健康牛相比,患病牛鼻咽部中的微生物多樣性降低,致病性病原菌如:牛支原體、溶血性曼氏桿菌、牛巴氏桿菌等相對豐度顯著升高。本試驗在不同地區患病牛鼻咽部中也檢測到以上3種致病性細菌,除此之外,還檢測到不動桿菌屬和金黃桿菌屬。研究發現,金黃桿菌感染常發生在慢性呼吸道疾病和免疫力低下的宿主中[22];弓清梅等[23]發現腦膜膿毒金黃桿菌會導致下呼吸道疾病的發生,且對絕大多數抗生素耐藥。產吲哚金黃桿菌的耐藥性也極其復雜[24]。不動桿菌是一種機會性致病菌,常引起肺部炎癥,其易感群體與金黃桿菌相似,該菌對市面上常用的抗菌素具有耐藥性,導致臨床治療困難,病死率較高[25]。以上結果表明,除常見的致病性細菌外,金黃桿菌屬和不動桿菌屬可能導致患慢性呼吸道疾病牛和長期使用抗菌素的牛場的繼發性感染。
本試驗對患BRD牛鼻咽真菌菌群進行了測序分析,發現與呼吸系統疾病相關的真菌屬,如:曲霉菌屬、假絲酵母屬和克柔氏菌屬等,其中,曲霉菌屬在慢性呼吸系統疾病中最為常見[13]。本研究3組樣品中均檢測曲霉菌屬,R3組中相對豐度最高,表明曲霉菌屬是R3組的主要致病性真菌。假絲酵母屬也是引起下呼吸道感染的真菌之一,在R1組中的相對豐度大于5%。陳太方等[26]在研究抗真菌藥物對下呼吸道感染的抑菌作用中發現假絲酵母屬檢出率是多種真菌中最高的,且具有一定的耐藥性。克柔氏菌屬在R1和R2組中的相對豐度均大于30%,該菌屬條件性致病菌,高豐度的克柔氏菌可導致支氣管肺炎的發生[27]。上述結果表明了在患呼吸系統疾病牛鼻咽微生物可檢測出致病性真菌,可能影響呼吸道疾病的發展,具體致病機制仍需進一步探究。
3.3 呼吸系統疾病牛鼻咽部菌群功能預測
鼻咽部微生物作為牛機體微生態系統的重要組成部分,微生物組與機體的互作對維持宿主生理穩態和機體健康至關重要。在相似的的環境中,盡管群落結果發生了巨大變化,但其功能仍然相對穩定[28]。研究者對牛瘤胃微生物及腸道微生物功能進行了分析,發現功能集中在氨基酸運輸和代謝、碳水化合物轉運及代謝的相關基因上[2]。大多數的牛呼吸道疾病微生物研究只分析了結構組成、相對豐度及其與疾病的相關性,很少針對微生物測序結果進行功能預測[8]。基于PICRUSt和FunGuild數據庫,本研究揭示了在患呼吸系統疾病的牛鼻咽部細菌類KEGG二級水平的通路中,R1組和R2組細菌類微生物導致代謝類的相關通路(氨基酸代謝、碳水化合物代謝、消化系統等)豐度增加,可能是由于細胞和結締組織破壞,與肺功能受損和全身炎癥相關[29]。其中消化系統相對豐度增加,可能與肺-腸軸通路有關[30]。R3組與其他2組的富集結果存在差異,功能基因豐度增加的通路主要包括疾病感染、遺傳信息呈遞和信號轉導等,這可能與呼吸系統疾病的發生、微生物在呼吸道中增殖及宿主-微生物互作有關。真菌類群功能預測結果顯示,3組樣品的上呼吸道中均存在動物病原菌富集及豐度增加,表明牛呼吸系統疾病存在致病性真菌感染的可能,為臨床治療提供了方向。
4 結論
本研究從患BRD牛鼻咽微生物出發,分析了鼻咽微生物中細菌、真菌的群落組成、相對豐度及功能預測。鑒定出牛鼻咽部致病性細菌,如:牛支原體、巴氏桿菌、曼氏桿菌、金黃桿菌及不動桿菌;致病性真菌,如:曲霉菌屬、假絲酵母屬和克柔菌屬,說明患呼吸系統疾病牛鼻咽微生物菌落組成豐富,且包含多種致病性微生物。
猜你喜歡
鐘表(2023年5期)2023-10-27 04:20:44
中華詩詞(2022年6期)2022-12-31 06:41:24
當代陜西(2021年21期)2022-01-19 02:00:26
中學生數理化(高中版.高考數學)(2020年1期)2020-02-20 13:23:44
經濟技術協作信息(2018年11期)2019-01-14 03:07:20
中國科技論壇(2017年7期)2017-07-25 08:49:53
制造技術與機床(2017年3期)2017-06-23 08:11:33
媽媽寶寶(2017年2期)2017-02-21 01:21:24
國際漢語學報(2016年1期)2017-01-20 08:21:20
中國中醫藥現代遠程教育(2014年22期)2014-03-01 04:32:55
