基于微生物組大數據搜索的疾病檢測
2021-09-06 18:37:50張玉鳳荊功超李勁華蘇曉泉
科學 2021年2期
張玉鳳 荊功超 李勁華 蘇曉泉
微生物是地球上最早進化形成的生命體之一,它們與后續進化的動植物以及人類等相互影響,形成各種各樣的復合體系。微生物在自然生態圈中幾乎無處不在,它們絕大多數并不是孤立的,而是以“微生物群落” (亦稱菌群)的形式共存。通常,也用“微生物組”(microbiome)來表示某個特定環境或者生態系統中全部的微生物及其遺傳信息。
人體微生物組被稱為人的“第二基因組”。 作為與生俱來又無處不在的“小伙伴”,微生物組與我們有著千絲萬縷的關聯[1]。隨著生物技術與大數據的發展,研究發現人體菌群與人的健康狀況的變化和疾病的發生發展密切相關,如炎癥性腸病[2]、Ⅱ型糖尿病[3]、結直腸癌[4]等疾病的發生都伴隨著腸道菌群的改變。微生物組檢測具有非侵入性、可量化性、可預警等優勢,例如,兒童的口腔菌群檢測僅需幾滴唾液,就可以提前預測齲齒的患病風險[5],使其在疾病識別、治療方案制定、預后評估等方面有著巨大的潛力。因此,基于微生物組的健康狀態檢測一直是精準醫學和大健康領域的熱點問題之一。那么,如何用菌群判斷和區分疾病呢?
微生物組疾病檢測的方法與不足
一個微生物群落中可能存在成百上千種不同的物種。因此,微生物組檢測首先用DNA測序和生物信息分析來獲取菌群的結構成分,也就是知道其中有什么微生物,含量比例是多少。之后,常規的做法是將某類疾病患者的菌群樣本和健康樣本進行對比,發現其中關鍵的差異物種作為生物標記(bio-marker),并根據標記的含量高低來判斷待檢樣本是否罹患該類疾病。然而,這類方法在適用廣泛性、疾病特異性、污染抵抗性等方面有著明顯的不足,從而阻礙了微生物組技術的進一步應用。
首先,生物標記法在疾病普適程度上限制大,易出現“漏診”問題。利用某類疾病樣本和健康樣本中具有差異的物種作為生物標記,檢測范圍也就僅限于該疾病類型,否則將無法正確地識別出樣本的狀態。然而在實際的檢測情況中,不同人群中同一疾病的生物標記物種往往不一致[6],甚至許多疾病沒有明確的標記。當樣本的疾病超出模型的檢測范圍時,很難綜合地判斷待測樣品是否健康。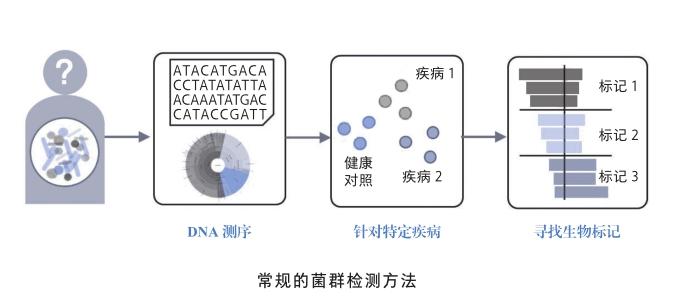
同時,生物標記法在檢測特異性上的缺陷會導致“誤診”問題。前期研究表明,已知的每一種生物標記均會與至少兩種不同的疾病存在關聯[7]。因此,生物標記的檢測范圍較大,涵蓋的疾病類型較多(例如5個以上)時,在區分不同疾病的過程中會容易出現誤判。
此外,生物標記容易受到外來“污染物”的干擾,從而影響檢測結果。由于我們身體上和生活的環境中幾乎所有可及之處都有微生物的存在,檢測的過程中可能會混入來自身體其他部位或者環境中的微生物,這些“污染物”會改變菌群成分分析中生物標記的相對含量,從而降低疾病識別的穩定性。
微生物組搜索引擎和大數據挖掘為疾病檢測提供新思路
隨著DNA測序成本的降低和微生物組研究的迅猛發展,菌群的測序數據正在以前所未有的速度累積。然而,每當新的微生物組數據產生時,受限于該領域大數據分析工具的匱乏,難以與原有的海量數據進行快速的比對,束縛了對新數據的認識與解讀。筆者團隊成功開發了微生物組搜索引擎(Microbiome Search Engine,以下簡稱搜索引擎),通過物種成分分類的動態索引機制和Meta-Storms比對算法[8],實現了微生物群落整體層面的高速搜索,從而在急速增長的已知數據中,迅速準確地找到與新樣本在群落結構上高度相似的匹配樣本,使得未知菌群的“溫故而知新”成為可能。
對于每一個新的微生物群落,利用搜索引擎在已有的數據庫中進行搜索,根據最佳的匹配度來計算的“微生物組新穎指數”(以下簡稱新穎指數),能夠量化出該樣本的新奇性和獨特性。新穎指數值越高說明樣本與先前已大規模采樣收錄的菌群相似性越低,也就越獨特。研究人員通過對2010—2017年產生的來自全球不同地域、不同環境的共100 000多例微生物組樣本進行追蹤分析,結果顯示人體菌群的新穎指數變化在全局范圍內是收斂性發展,且已經呈現接近飽和的狀態[9],而自然環境菌群的收斂性卻不明顯。人體微生物組的這種收斂性也稱為 “搜索邊界效應”,即雖然人與人之間菌群結構和組成差異很大,但從整體尺度來看,其變化總在一定的范圍之內。
當進一步縮小范圍,健康的人體微生物組是不是也存在類似的“搜索邊界效應”,使得健康菌群的新穎指數總在一定正常范圍之內,而疾病菌群明顯高于正常水平,從而利用搜索的策略來實現對非正常狀態菌群的篩選和識別。研究人員再次利用多個地區超過15 000例正常人群的微生物組作為基準,發現多種不同疾病樣本的新穎指數均要顯著高于健康樣本,從而驗證了健康微生物組的“搜索邊界效應”,并為疾病檢測提供了前提。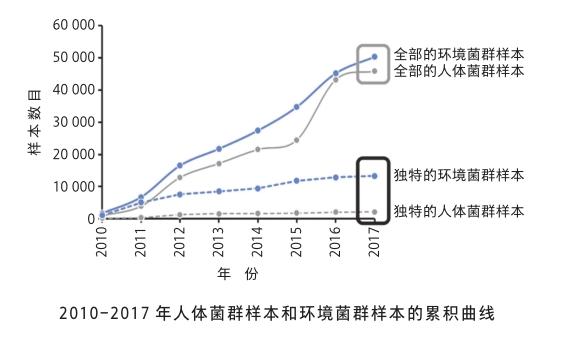
基于大數據搜索的微生物組疾病檢測
與現有的生物標記法相比,基于搜索的疾病檢測不再依賴疾病相關的特定標識微生物,而是利用待測菌群的整體組成結構與已有數據的相似度來實現檢測[10]。此方法分為兩步:第一步,計算待測樣本相對于健康數據庫的新穎指數,根據其異常程度即可評估其是否健康;第二步,與疾病數據庫進行比對,根據最佳匹配結果即可識別具體的患病種類,從而依次回答“是否健康”和“哪種疾病”這兩個問題。
經過3000余例人體腸道微生物組樣本的檢測表明,針對炎癥性腸病(inflammatory bowel disease, IBD)、結直腸癌(colorectal cancer, CRC)、艾滋病毒感染(human immunodeficiency virus,HIV)和腸腹瀉病(enteric diarrheal disease,EDD)這4種疾病,搜索引擎在回答“是否健康”和“哪種疾病”這兩個問題上的準確率均超過75%,顯著高于目前常用的生物標記法,從而有效地降低了“漏診”和“誤診”的可能。此外,由于炎癥性性病和腸腹瀉病具有大量相同的生物標記,導致生物標記法容易產生混淆,降低了對二者的區分度和識別率,從而產生了“短板效應”,而搜索引擎對其中每一種疾病的檢測準確性都較為一致。
基于搜索的檢測方法對微生物污染具有較強抵抗性
一般來講,科學實驗數據的采樣、封存、測序等過程由專業的操作人員按照標準工作流程來完成,會在極大程度上降低污染的可能性。而當未來菌群檢測逐漸普及后,菌群的采樣會由用戶或患者自行來完成。例如,當我們通過棉簽涂抹手部來獲得皮膚菌群時,可能會難以避免地混入其他身體部位或者生活環境當中的污染微生物,從而干擾檢測結果。為了測試不同檢測方法對污染物的抵抗性,研究人員在測試數據中分別混入5%~20%不同比例的室內環境微生物作為“污染”。結果顯示,即便在最高的污染率下,搜索引擎仍具有71%以上的良好準確度。但由于高污染會顯著改變群落中物種的相對比例,生物標記法檢測結果受影響較大,平均識別率已然驟降到了60%左右。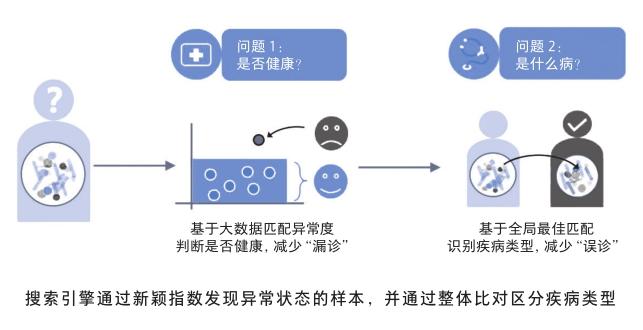
以多角度、大數據分析為特征的微生物組研究時代已經來臨[11,12]。得益于交叉學科研究,配合微生物組高通量測序和高性能計算方法學等方面迅猛的進步,以大數據驅動為主的微生物組創新研究模式將會為微生物組技術在人體健康領域的研究和臨床應用帶來突破性新手段和新思路,深化對共生菌群和人類健康之間互作關系的認識,造福社會,造福人類,造福自然。
[1]Turnbaugh P J, Ley R E, Hamady M, et al. The human microbiome project: exploring the microbial part of ourselves in a changing world. Nature, 2007, 449(7164): 804-810.
[2]Halfvarson J, Brislawn C J, Lamendella R, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nature Microbiology, 2017, 2(5): 17004.
[3]Qin J J, Li Y R, Cai Z M, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature, 2012, 490(7418): 55-60.
[4]Wirbel J, Pyl P T, Kartal E, et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specifi c for colorectal cancer. Nature Medicine, 2019, 25(4): 679-689.
[5]Teng F, Yang F, Huang S, et al. Prediction of early childhood caries via spatial-temporal variations of oral microbiota. Cell Host & Microbe, 2015, 18(3): 296-306.
[6]Duvallet C, Gibbons S M, Gurry T, et al. Meta-analysis of gut microbiome studies identifi es disease-specifi c and shared responses. Nature Communications, 2017, 8(1): 1784.
[7]Jackson M A, Verdi S, Maxan M E, et al. Gut microbiota associations with common diseases and prescription medications in a population-based cohort. Nature Communications, 2018, 9(1): 2655.
[8]Su X, Xu J, Ning K. Meta-Storms: Efficient search for similar microbial communities based on a novel indexing scheme and similarity score for metagenomic data. Bioinformatics, 2012, 28(19): 2493-2501
[9]Su X, Jing G, McDonald D, et al. Identifying and predicting novelty in microbiome studies. mBio, 2018, 9(6): e02099-18.
[10]Su X, Jing G, Sun Z, et al. Multiple-disease detection and classification across cohorts via microbiome search. mSystems, 2020, 5(2): e00150-20.
[11]Su X, Jing G, Zhang Y, et al. Method development for crossstudy microbiome data mining: challenges and opportunities. Computational and Structural Biotechnology Journal, 2020, 18: 2075-2080.
[12]Taroncher-Oldenburg G, Jones S, Blaser M, et al. Translating microbiome futures. Nature Biotechnology, 2018, 36(11): 1037-1042.
關鍵詞:微生物組 大數據 搜索引擎 疾病檢測 生物信息 ■
猜你喜歡
新聞世界(2016年10期)2016-10-11 20:13:53
科技視界(2016年20期)2016-09-29 10:53:22
中國記者(2016年6期)2016-08-26 12:36:20
中國衛生(2015年12期)2015-11-10 05:13:38
警察技術(2015年3期)2015-02-27 15:37:09
新疆大學學報(自然科學版)(中英文)(2014年2期)2014-11-06 07:49:12
技術經濟與管理研究(2014年11期)2014-03-11 17:02:44
百科知識(2012年11期)2012-04-29 08:30:15
計算機應用文摘(2009年17期)2009-04-29 00:44:03
