基于Rietveld精修方法解析Bi2O3的原子熱振動
2021-09-22 07:32:14王丹丹劉澤朋
人工晶體學報 2021年8期
王丹丹,劉澤朋,香 蓮
(內蒙古民族大學數理學院,通遼 028043)
0 引 言
Bi2O3是氧離子(O2-)導電體,它是一種先進的功能材料,也是最重要的鉍化合物之一。近年來,因其具有大的能帶隙、折射率、介電常數以及顯著的光電導和光致發光等光學和電學性質而被廣泛應用于電子陶瓷材料、電解質材料、光電材料、傳感器、微電子元件、高溫超導材料、催化劑、鐵電材料等各領域中,同時還用于化學試劑、鉍鹽、防火材料、高折光率玻璃、核工程玻璃制造和核反應堆燃料等方面[1]。Polat等[2]制備了Gd2O3和Lu2O3雙摻雜Bi2O3化合物,表明了穩定的δ-Bi2O3相樣品可用作固體氧化物燃料電池的電解質材料。丁鵬等[3]制備了Bi2O3-Ni2O3納米粉末,經750 ℃焙燒的光催化劑對苯光催化降解活性最高。Hakimi等[4]通過非水溶膠-凝膠路線合成Bi2O3-Al2O3納米復合材料,顯示出比Al2O3更高的光催化活性。Viruthagiri等[5]用一種新的化學沉淀方法來制備不同濃度的鈷摻雜Bi2O3納米顆粒,表現出優異的結構和光學性質。Raju等[6]研究了ZnO-Bi2O3-Yb2O3基壓敏陶瓷,提高了壓敏電阻組件在侵入納秒瞬態下從非導電態轉變到導電態的轉變速度。徐東等[7]制備了摻Lu2O3的ZnO-Bi2O3基壓敏陶瓷樣品,具有比較理想的綜合電性能。熊智慧等[8]采用第一性原理雜化泛函HSE06方法對Fe摻雜α-Bi2O3的電子結構和光學性質進行了計算研究,為Fe摻雜α-Bi2O3在光催化領域中的應用提供了理論依據。Huang等[9]通過溶劑熱制備和后退火處理來修飾Eu3+摻雜的鉍氧化物的相形成、形態特征和光能轉化,提出了光能轉換機理來討論發光性質、衰變壽命和多價離子。三氧化二鉍(Bi2O3)有七種晶相,分別為單斜α相(α-Bi2O3)、四方β相(β-Bi2O3)、體心立方γ相(γ-Bi2O3)、面心立方δ相(δ-Bi2O3)、正交ε相(ε-Bi2O3)、三斜ω相(ω-Bi2O3)和六方η相(η-Bi2O3)。其中最穩定的結構為α相和δ相,亞穩定結構為β、γ、ε和η相,而ω相因極不穩定而存在爭議[10]。本研究通過X射線對粉末晶體Bi2O3進行衍射實驗,利用Rietveld解析方法中的RIETAN-2000程序[11]對實驗結果進行全譜擬合晶體結構精修,得到原子熱振動各向同性因子B。B是與材料熱性質相關的重要參數,利用它可計算出德拜-勞厄因子,也可推導出原子間熱振動相關效果μ的值,并進一步計算出比熱和色散譜[12],原子熱振動大小的B值與溫度有關,且隨溫度增高而增大[13-14]。用最大熵方法(maximum entropy method, MEM)解析方法通過Practice Iterative MEM Analyses(PRIMA)模塊和Vi-sualization of Electron/Nuclear Densities(VEND)模塊[15-19]進行等高電子密度分布可視化,確定晶體結構和原子的位置。
1 Rietvld精修和MEM解析
1.1 Rietvld精修
Rietveld精修是由設在荷蘭的Petten反應堆中心的研究員Rietveld在1967年用中子多晶體衍射數據精修晶體結構參數時提出的一種數據處理新方法——多晶體衍射全譜線形擬合法。1977年,Malmros和Thomas首次將這一方法應用到X射線領域。Rietveld全譜擬合是以一個晶體結構模型為基礎,利用它的各種晶體結構參數及一個峰形函數計算一張在2θ范圍內的、理論的多晶體衍射譜,將此計算譜與實驗測得的衍射譜比較并修改,反復進行多次,以使計算譜和實驗譜的差最小(最小二乘法),擬合的目標是整個衍射譜的線形,擬合范圍是整個衍射譜,不是個別衍射峰。整個衍射譜是各衍射峰強度分布的疊加,故衍射譜上某點 (2θ)i處的實測強度Yi表示為
Yi=Yib+∑kYik
(1)
式中:Yib為本底強度。在作Yik加和時,需設定每個衍射峰的延展范圍,定義為該峰的半峰寬(FWHM)的若干倍,如5倍或7倍等。這樣可以知道在某個(2θ)i處的Yi是由哪幾個衍射貢獻的。根據一定的晶體結構模型及峰形函數,可以計算衍射譜上各(2θ)i處的衍射強度Yic(下標c表示為計算值)。改變各結構參數或峰形參數,可改變Yic。用非線性最小二乘法使Yic擬合實測的高分辨、高準確的數字多晶體衍射譜上的各Yio(下標o表示實測值),也即下式之中M最小:
M=ΣiWi(Yio-Yic)2
(2)
式中:Wi=1/Yi為基于計數統計的權重因子;Yio為實測值,在反復循環中不變;Yic是根據模型計算的,在每次參數修改后,此值均要改變。
為了判別精修中各參數的調整是否合適,設計出一些判別因子,簡稱R因子。
RI=Σ|Iko-Ikc|ΣIko
(3)
(4)
式中:Iko表示積分強度的實測值;Ikc表示積分強度的計算值。這兩個因子是由衍射峰的積分強度計算出的,強烈依據于結構模型,是能判斷結構模型是否正確的最有價值的R因子[20]。
1.2 最大熵方法
MEM起源于Jaynes引入的統計力學領域的“最大熵原理”,原本是物理學中的一個概念,用來反映某一系統的混亂程度,后被用于數學中來描述一個信息量,即信息熵,是指在推斷未知分布時,只考慮已知信息,而不隨意加入主觀信息,信息量越大,則熵也越大[21-22]。在熱力學的相關研究中表征系統的混亂程度和無序度。在信息科學中,熵是對信息的不確定度的一種測量,用來作為一個系統的信息含量的量化指標[23-24]。此方法在地球物理學科中各種數據處理中獲得了跨時代的成功,已在許多科學和技術領域中采用,例如光譜分析、圖像恢復、數學和物理學等,本研究將其應用到晶體結構分析上。MEM是根據有限的信息,使用信息熵推導出統計性的最準確結論的方法,1967年由J.P.伯格所提出[25]。這個方法中,要求實驗中得到的信息吻合這個限制條件下得到的信息熵的最大解。最大熵原理的含義就是,在已有的條件約束下,不添加任何人為假設,使熵達到最大,即混亂程度最高[26]。對于衍射實驗中,決定電子-原子核密度時,推定觀察到的結構因子在誤差范圍內的密度分布盡量取分散值,對于沒有被測定的高角度領域部分的結構因子推定為不等于零的結構因子。

(5)
(6)
對于τj的初期值,就是熵的最大信息狀態,即在體積V的單位胞中通常采用密度均衡分布的模型。在X射線衍射的情況下,如果F(000)是000反射的構造因子(等于單位胞中總電子數),那么N個像素全部設定為F(000)/V。
在衍射實驗領域里MEM解析是Dilanian和泉富士夫兩人開發建立的電子-原子核密度的可視化(visualization of electron/nuclear densities and structures, VENUS)程序系統。VENUS可以在三維(3D)可視化、渲染和操縱晶體結構以及各種物理量(例如電子/核密度)中發揮作用,不僅可以通過X射線和中子衍射來確定,還可以通過電子結構計算來確定,能夠防止實驗方法與理論方法之間的相分離[27]。
2 實 驗
本研究使用的是天津市光復精細化工研究所的純度不低于99.99%的Bi2O3粉末晶體。
首先使用型號為BT-1600的圖像顆粒分析系統對Bi2O3粉末晶體進行顆粒分析,BT-1600圖像顆粒分析系統包括型號為生物、金相、體視顯微鏡(三目),最大光學放大倍數為1 600倍的光學顯微鏡、500萬像素的型號為RZ500C數字CCD攝像頭、數據采集與處理的BT-1600軟件、windows系統電腦以及彩色噴墨打印機,其重復性誤差<3%(不包含樣品制備因素造成的誤差)。其原理為通過對顆粒數量和每個顆粒所包含的像素數量的統計,計算出每個顆粒的等圓面積和等球面積,從而得到顆粒的等圓面積直徑、等球體積直徑以及長徑比等,再對所有的顆粒進行統計,從而得到粒度分布等信息,以微米為單位。其實驗結果如表1和圖1所示,實驗結果表明,Bi2O3粉末晶體呈現片狀,最大粒徑為91.28 μm,最小粒徑為0.60 μm,平均粒徑為 9.42 μm,比表面積為0.092 m2/g。

圖1 Bi2O3粒度分析的形狀Fig.1 Shape of Bi2O3 particle size analysis

表1 室溫下Bi2O3的粒度分析Table 1 Particle size analysis of Bi2O3 at room temperature
使用型號為D8 FOCUS的X射線衍射儀,對粉末晶體在室溫條件下進行了衍射實驗。輻射源為Cu Kα,其波長λ為0.154 nm,掃描范圍10°~90°,步長0.02°,每步計數時間為3 s。實驗結果如圖2所示。

圖2 室溫下X射線衍射實驗結果Fig.2 X-ray diffraction experiment results at room temperature
3 結果與討論
室溫條件下α-Bi2O3屬于單斜晶系,其空間群為P21/c,每個晶胞含有8個Bi原子和12個O原子[28-29]。如圖3為Bi2O3的晶體結構模型,利用RIETAN-2000程序對Bi2O3晶體結構進行了Rietveld精修,精修圖譜如圖4所示,實線表示計算值,點線表示 X 射線衍射實驗值,短線表示峰的位置,下方的波動線是兩者的差值。圖中斷開部分因此區間內出現了幾處γ-Bi2O3的衍射峰[30]而將其刪除。從精修結果可以看出,Bi2O3的實驗圖譜和計算值圖譜均匹配得很好。
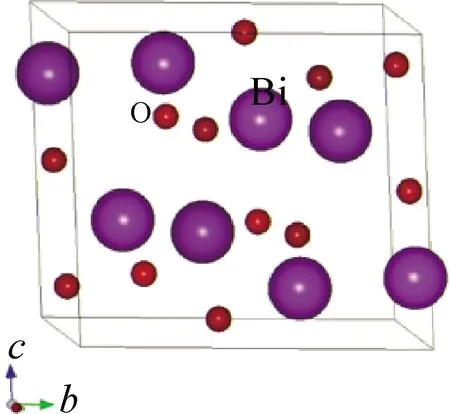
圖3 Bi2O3晶體結構模型Fig.3 Bi2O3 crystal structure model

圖4 Bi2O3精修圖譜Fig.4 Bi2O3 refined map
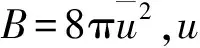

表2 Bi2O3晶體結構參數Table 2 Bi2O3 crystal structure parameters
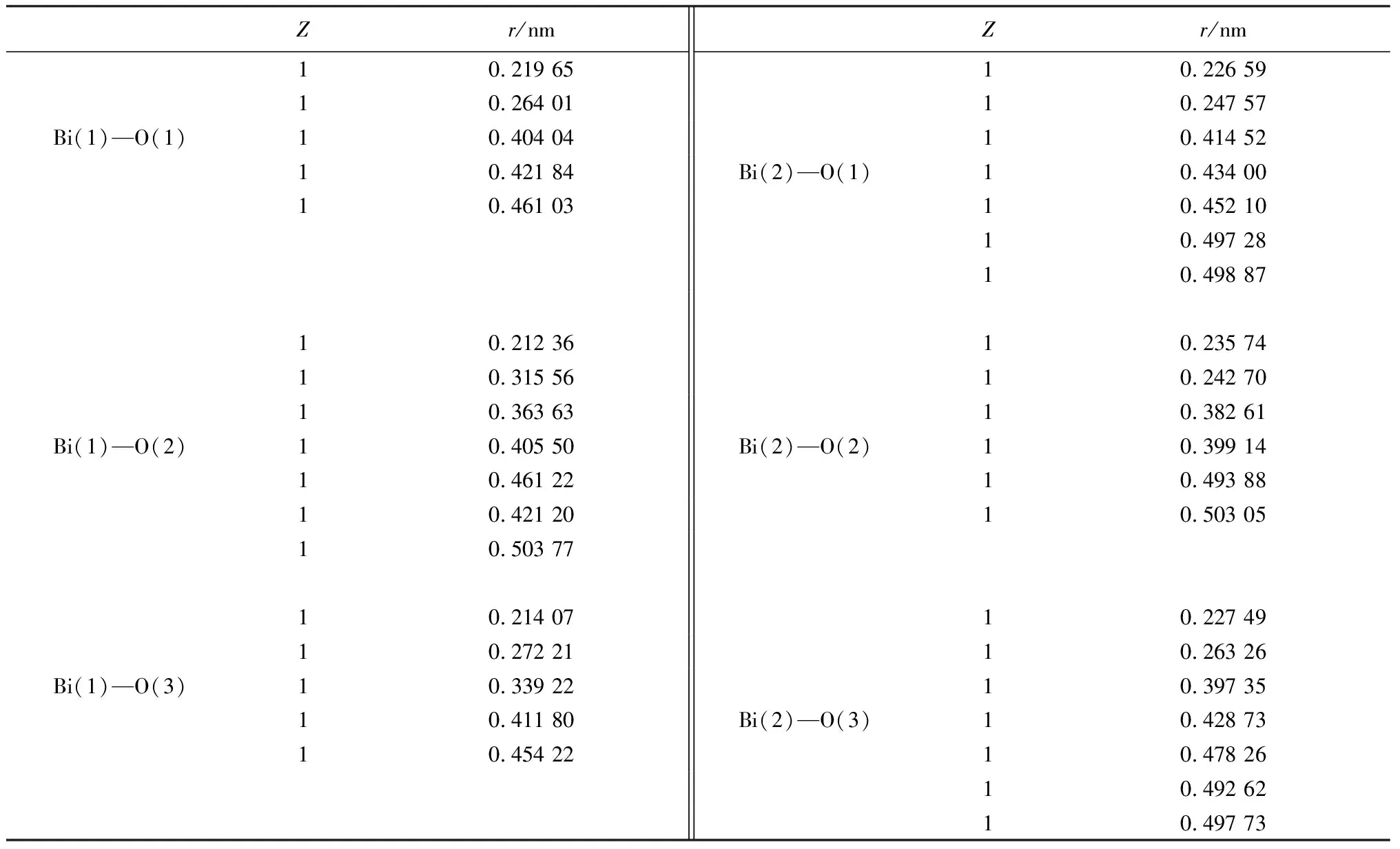
表3 Bi2O3原子配位數Z和原子距離rTable 3 Bi2O3 atomic coordination number Z and atomic distance r
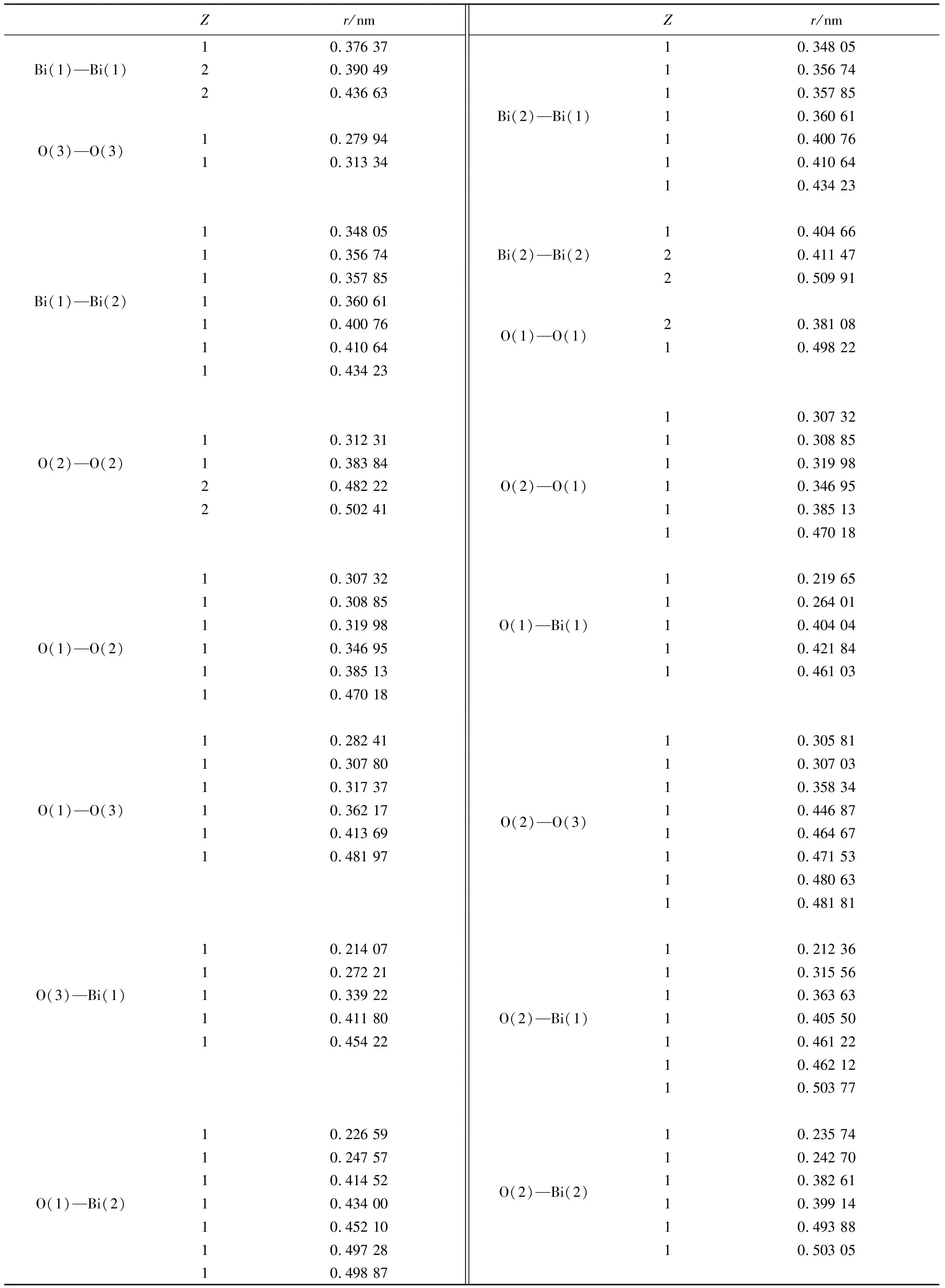
續表

續表
衍射強度公式中e-2M被稱為德拜-沃勒因子(Debye-waller factor),其中M=B(sinθ/λ)2。雖然,B的數學公式已報道,但是通過公式計算極其困難,利用晶體結構精修用的Rietveld程序軟件可以直接得到B的準確值。
圖5和圖6是采用MEM解析方法的PRIMA模塊和VEND模塊計算的3D(立體)和2D(平面)等高電子密度可視化分布圖譜。圖5、6所示的3D(立體)和2D(平面)等高電子密度分布圖譜里的電子密度分布是球狀分布,擴散面積小,這進一步說明了在室溫附近各原子作各向同性的熱振動。圖5顯示的3D等高電子密度分布圖與圖3的晶體結構模型非常符合,證明了初步建立的晶體結構模型是正確的。圖6為(100)晶面的2D電子密度分布圖,從立體和平面圖可以看出Bi原子的確切位置,而O原子幾乎看不見,這是由于Bi原子的原子序數為83,O原子的原子序數為8,X射線衍射中衍射強度與原子散射因子f有關,而原子散射因子f的大小與原子序數有關,原子序數越大原子散射因子f越大,所以重原子很容易被X射線觀察到,而輕原子不容易被觀察到。
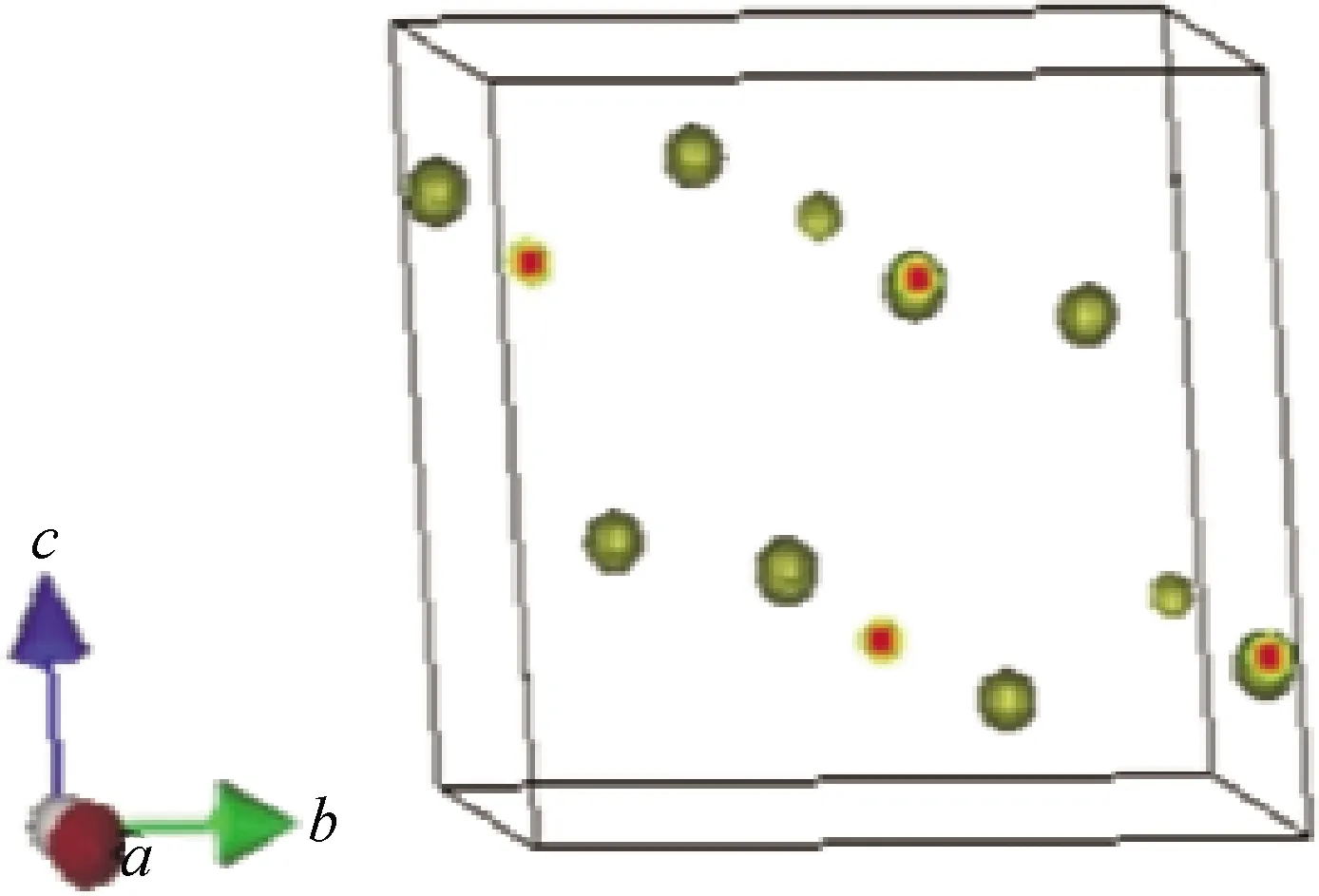
圖5 3D電子密度可視化圖譜Fig.5 3D electron density visualization map
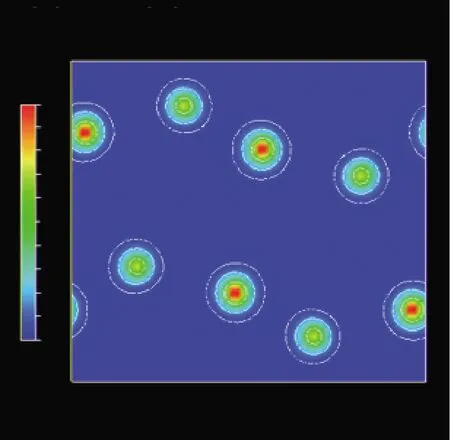
圖6 (100)晶面的2D電子密度可視化圖譜Fig.6 2D electron density visualization map of the (100) crystal plane
4 結 論
本研究首先對Bi2O3粉末晶體進行顆粒分析,發現Bi2O3粉末晶體呈現片狀,最大粒徑為91.28 μm,最小粒徑為0.60 μm,平均粒徑為 9.42 μm,比表面積為0.092 m2/g。然后對Bi2O3粉末的晶體結構進行了精修,得到了原子熱振動各向同性因子B的大小,各原子Bi(1)、Bi(2)、O(1)、O(2)和O(3)的原子熱振動各向同性溫度因子分別為0.004 938 nm2、0.004 174 nm2、0.007 344 nm2、0.007 462 nm2和0.007 857 nm2。B是與材料熱性質相關的重要參數,利用它可計算出德拜-勞厄因子,也可推導出原子間熱振動相關效果μ的值,并進一步計算出比熱和色散譜。所以進一步的研究目標是結合定性和定量分析探究離子導電率和原子熱振動之間的關系。通常,原子熱振動各向同性溫度因子B的計算比較復雜,可以通過X射線衍射實驗與晶格精修方法結合直接得出。
猜你喜歡
科學大眾(2023年17期)2023-10-26 07:39:14
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
天天愛科學(2020年6期)2020-09-10 07:22:44
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
數學物理學報(2017年6期)2018-01-22 02:26:40
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
