調節破骨細胞功能的相關信號分子的研究進展
2021-10-08 10:07:40趙常紅李世昌李沛鴻翟晶
中國骨質疏松雜志 2021年9期
關鍵詞:信號
趙常紅 李世昌 李沛鴻 翟晶
1.西北師范大學體育學院,甘肅 蘭州 730030 2.華東師范大學體育與健康學院,上海 200241 3.甘肅省人民醫院,甘肅 蘭州 730000
破骨細胞是通過分泌組織蛋白酶K(cathepsin K, CtsK)、H+、Cl-和基質金屬蛋白酶(matrix metalloproteinases, MMPs)來發揮蝕骨作用的一種大型多核細胞。破骨細胞前體細胞在M-CSF刺激下與相應受體c-Fms結合,并誘導RANK的表達,促進破骨細胞的生成[1]。通過RANKL刺激相應受體驅動下游信號因子,導致破骨細胞生成。本文通過近年來調控破骨細胞的分化和活性信號通路進行研究,探究其復雜關系,為防治骨質疏松癥的藥物研發提供可靠的分子生物學機制依據[2]。
1 RANKL/RANK/OPG信號通路
作為調控破骨細胞的主要信號通路,RANKL/RANK/OPG最為重要[3]。RANKL是TNF超家族的一種多肽II型跨膜蛋白,由成骨細胞產生,水解后以可溶性形式存在于細胞表面,能激活破骨細胞生成和遷移,增加骨吸收能力,然后周邊的成骨細胞立即聚集填充骨陷窩形成新的骨表面,這就形成骨的重塑[4]。
OPG是一種可溶性蛋白,也由成骨細胞產生是RANKL的誘餌受體,能競爭地阻止RANKL與受體RANK的結合,能阻止破骨細胞的形成與蝕骨能力的增加[5]。OPG也屬于TNFR超家族,也包含4個CRD,并且在CRD2和CRD3之間有一個鉸鏈區域,可以結合到RANKL。因此,OPG通過競爭性地阻止RANKL和RANK的結合,調控破骨細胞的產生與活性,進而調節骨的構建與重塑,與RANK競爭性調節破骨細胞分化與活性,在骨重塑中具有十分重要的作用[6]。
RANK是RNAKL的信號受體,屬于腫瘤壞死因子受體(TNFR)超家族分子的一種I型跨膜蛋白,由4個富含半胱氨酸的結構域(CRD)組成。RANK信號的激活可促進一些特異基因的表達,有利于促進破骨細胞分化、成熟,增加破骨細胞存活時間,激活破骨細胞骨吸收能力。RANK信號也能激活ERK-1激酶 ,ERK信號通路可促進破骨細胞形成[7]。RANK誘導的下游信號變化是判別破骨細胞活化的重要標識基因,尤其是TRAF6,RANKL與 RANK結合后,通過TRAF6可以將信號釋放放大到下游 NFκB、MAPK、Src-AKT、Src-Ca2+信號通路中去,促進破骨細胞分化、成熟和蝕骨能力增加。
2 NF-κB信號通路
NF-κB是一個轉錄因子家族,包括RelA(也稱為p65)、p50、p52、RelB和c-Rel,對破骨細胞的形成是必不可少的[8]。NF-κB p50和p52蛋白的共同表達,破骨細胞才能成熟發揮蝕骨功能。在破骨細胞分化過程中RANKL-RANK信號傳遞到下游通路主要通過IKKβ及其經典的NF-κB信號通路[9]。在RANKL刺激后,NF-κB、AP-1、NFATc2、ATF4和Jdp2被招募到NFATc1基因的啟動子區,共同誘導NFATc1,這種轉錄因子的表達在RANKL被自身擴增機制刺激后大大上調,增強破骨細胞的生成。NF-κB p50和p52亞基的缺失導致破骨細胞的缺乏導致骨質疏松,隨后觀察到NF-κB對于RANKL和其他破骨細胞因子誘導的RANK表達的破骨細胞前體分化為TRAP+破骨細胞至關重要[10]。c-Fos和活化T細胞的核因子、NFATc1以及抑制NFATc1信號的抑制因子,也通過NF-κB信號蛋白、TRAF3和p100以及c-Fos/NFATc1信號轉導抑制因子IRF8激活NF-κB信號轉導,使破骨細胞皺褶的邊緣膜下形成鹽酸,溶解骨的礦物質成分,組織蛋白酶K被分泌以降解基質[11]。
3 MAPK /ERK信號通路
MAPK屬于蘇氨酸/絲氨酸蛋白激酶家族,由胞外信號調節激酶(Erk1/2)、p38-MAPKs(α/β/γ/δ)、c-Jun N末端激酶(JNK1、2、3)等組成。p38-MAPKs的激活在 RANKL誘導破骨細胞前體細胞向破骨細胞分化的過程中起到了重要作用。p38α在破骨細胞中表達最為豐富,這種信號分子的敲除導致破骨細胞的發生受損。單核細胞和巨噬細胞MAPK14f/fLysM-Cre小鼠中,p38被特異性地刪除的小鼠顯示出破骨細胞骨吸收減少,骨量增加[12]。很多研究表明,除了MAPK-JNK-AP-1信號外,還有ERK5、ERK7和ERK8也參與了RANKL對破骨細胞的分化調節,MAPK和Akt級聯激活受M-CSF與其受體影響,從而使破骨細胞存活[13]。
ERK激活是成熟破骨細胞存活的核心,最近的研究表明p38 mapk-CREB通路在RANKL介導的破骨細胞分化中起重要作用。p38 MAPK抑制劑抑制TNF-α或RANKL誘導的CREB磷酸化,TNF-α或RANKL通過CREB磷酸化調節c-Fos和NFAT-c的表達,誘導破骨細胞分化[14]。最近的一項研究表明,Ambn通過抑制p38 MAPK-CREB磷酸化和下調c-Fos-NFAT c1軸來抑制RANKL誘導的破骨細胞分化[15]。p38 mapk在MAPK激酶激酶6(MKK6)激活后被激活,活化C激酶1受體(RACK1),在RANKL的反應下促進MKK6-p38 MAPK信號傳導,并參與破骨細胞分化,ERK1/2和p38 mapk-CREB通路在骨穩態中起重要作用[16]。
4 M-CSF 信號通路
眾所周知,單核/巨噬細胞系通過巨噬細胞集落刺激因子(M-CSF)的作用,是通過其受體c-fms誘導破骨細胞分化。GM-CSF和M-CSF是維持巨噬細胞數量和功能的重要因子。IL-34由成骨細胞產生,識別M-CSF受體(c-fms),從而促進破骨細胞的分化,如圖1[17]。M-CSF通過與其受體 c-fms結合,誘導受體胞質端的七個酪氨酸殘基進行磷酸化,在M-CSF誘導吞噬細胞和破骨細胞運動中,PI3K發揮非常重要作用。PI3K至少有3種不同的細胞表面受體下游效應子,包括 M-CSF受體、αvβ3以及 RANK。M-CSF能激活PI3K影響破骨細胞存活,也能調節破骨細胞肌動蛋白重塑,導致抑制膜皺褶、肌動蛋白環以及骨坑的形成。抑制PI3K的表達,既能有效抑制 RANKL及 M-CSF誘導的破骨細胞分化,也能降低其蝕骨能力。在 M-CSF/αvβ3整合素以及 DAP12-Syk信號通路下游,破骨細胞中調節細胞骨架重排的關鍵效應蛋白是 Vav3。Vav3-/-破骨細胞雖能正常分化,但不能形成胞足或密封圈,缺乏蝕骨能力[18]。
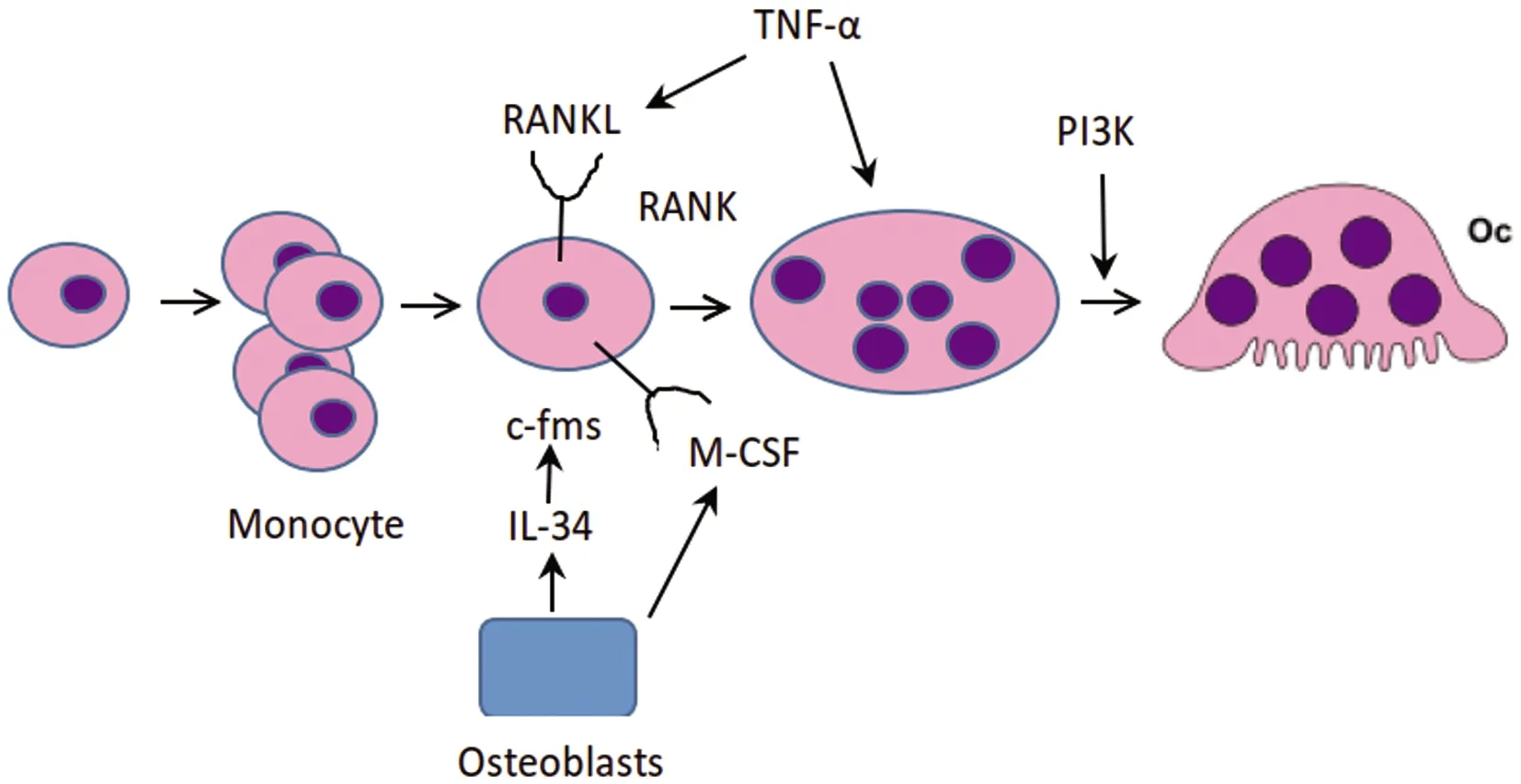
圖1 M-CSF調控破骨細胞生成示意圖Fig.1 Schematic diagram of osteoclast formation regulated by M-CSF
5 Ca2+信號通路
破骨細胞中的Ca2+信號是細胞分化、骨吸收和基因轉錄等多種細胞功能所必需的。研究發現,跨膜蛋白64(Tmem 64)是破骨細胞發生過程中Ca2+振蕩的調節因子,Tmem 64缺乏可顯著降低RANKL誘導的Ca2+振蕩,導致Ca2+/鈣調蛋白依賴性蛋白激酶(CaMK)IV和線粒體ROS減少,這兩個都有助于實現破骨細胞形成所需的CREB活性[19]。Aβ增強NF-κB活性和IκB-α降解,激活ERK磷酸化,刺激鈣振蕩,從而導致破骨細胞活化過程中NFATc1表達上調,且激活轉錄因子ATF3參與鈣信號傳導激活破骨細胞的分化和活性[20]。調控Ca2+攝入主要由間質相互作用分子(stromal interaction molecule,STIM)及 Orai1Ca2+釋放激活型鈣調節子完成。STIM-1、2是一種多域、單通的跨膜蛋白,參與感知細胞內Ca2+水平的變化,并將細胞信號轉導給Orai1通道蛋白,允許Ca2+內流。OCs中幾種Ca2+通道的表達,發現STIM1在OCs早期高表達,TRPV4在OCs晚期高表達,且STIM1和TRPV4分別作為OCs分化早期和晚期鈣通道可以作為破骨細胞發生或骨吸收的標志物[21]。研究發現,骨保護素通過Ca2+信號抑制破骨細胞分化和骨吸收,P2X7R-Ca2+-NFATc1信號通路在骨保護素誘導破骨細胞粘附結構損傷中起關鍵作用[22]。超過 24 h以上的RANKL刺激能使破骨細胞前體細胞發生Ca2+振蕩,長時間低水平的Ca2+信號能激活NFAT,促進破骨細胞形成[23]。
6 Src、Akt信號通路
編碼非受體酪氨酸激酶c-Src基因的缺失使成熟破骨細胞活動性降低,皺褶邊緣及骨吸收的相關細胞骨架異常,不能有效發揮蝕骨功能。Src誘導Cox促進破骨細胞線粒體內提供高水平ATP,調節破骨細胞骨吸收活性。巨核細胞相關酪氨酸激酶(Matk),一種有效的c-Src抑制劑,對破骨細胞有抑制作用[24]。研究發現,RACK1-c-Src軸作為破骨細胞功能的關鍵調節器的作用[25]。Pkn3以Wnt5a-Ror2信號依賴方式與c-Src和Pyk2結合,從而增強破骨細胞c-Src的激酶活性,促進破骨細胞的骨吸收活性[26]。c-Src-/-小鼠破骨細胞外圍胞足蝕骨帶缺失,表現出粘附、延展以及遷移上的缺陷,不具有蝕骨能力[18]。Src蛋白一般與 TRAF6結合,誘導破骨細胞存活、骨架重排和遷移[8]。
研究證明,Akt信號調節破骨細胞的融合,Akt抑制可通過降低RhoA活性和增強Akt/GSK3β/NFATc1信號傳導,觸發破骨細胞數量和活性的急劇增加[27]。已有研究證明,殼寡糖能通過激活AKT促進破骨細胞的分化。T63預處理顯著降低了Akt活性,抑制破骨細胞骨吸收,水蘇堿(STA)抑制Akt信號傳導,從而抑制活化NF-κB和NFATc1的活性,可減少破骨細胞數量并減輕骨量丟失[28]。苦皮藤醇(PIC)是一種天然的有機多酚二苯乙烯化合物,廣泛存在于多種植物中,可通過抑制MAPK、NF-kB和AKT信號通路抑制RANKL誘導的破骨細胞發生和骨吸收,并促進成熟破骨細胞凋亡[29]。CAL-10對RANKL誘導的Akt-c-Fos/NFATc1信號級聯抑制破骨細胞分化、遷移[30]。
7 PKC信號通路
PKC通路是破骨細胞重要的抑制性第二信使。研究發現,蛋白激酶 C(protein kinase, PKC)能調控破骨細胞分化與功能,如非典型 PKC(aPKC)支架蛋白 p62的突變會引起 Paget骨病,該病是一種破骨細胞異常激活導致的細胞功能障礙遺傳性疾病,且RANK信號誘導形成aPKC、TRAF6及p62復合物,有利于破骨細胞形成[13]。研究已經發現,PKCβ通過參與M-CSF和RANKL的ERK信號通路,參與破骨細胞的形成和功能的調節,揭示了細胞外酸化通過PKC依賴性途徑提高破骨細胞存活率,增加骨量丟失[31]。研究還發現p62相關的PKCζ在OCs過度活躍狀態和NF-κB活化中的重要作用[32]。PKC-d的藥物抑制和基因消融會損害體外破骨細胞骨吸收,破骨細胞前體的遷移依賴于通過整合素avβ3繞過RhoA和Rac1介導的PI3K/PKCa-PKCd信號傳導,而成熟破骨細胞的遷移依賴于整合素avβ3介導的PI3K/PKCaPKCd/RhoA-Rac1信號軸[33]。PKC-δ也是骨代謝的重要調節因子,在破骨細胞中PKC-δ的消融導致雄性小鼠骨小梁和皮質骨體積增加,而雌性小鼠則沒有觀察到骨量表型。伴隨著破骨細胞數量和表面積的減少,組織體內PKC蛋白水平以及破骨細胞的形成和吸收減少,是雄性特有的方式[34]。
8 Ig-like 受體信號通路
Ig-like的兩個受體接頭蛋白DAP12和FcRγ,表達在NK和骨髓細胞膜上,并通過ITAM結構域傳遞信號,這條信號通路是RANK的共調信號,Ig-like受體在破骨細胞分化中起重要作用。研究發現,細胞表面ITIM含有Ig樣受體調控破骨細胞形成,抑制性Ig樣受體招募Src同源2結構域的酪氨酸磷酸酶1(SHP-1)在破骨細胞前體細胞上表達,并在破骨細胞的發育中起調節作用,調節這些調節受體可能是控制各種骨骼系統疾病和炎癥性關節炎[35]。人破骨細胞相關受體(OSCAR)是一種免疫球蛋白(Ig)樣膠原受體,在破骨細胞發生過程中對破骨細胞上調,并在多種髓樣細胞中表達。此外,CLP肽作為破骨細胞生成的抑制劑,發現需要40個氨基酸的肽段才能在體外充分抑制破骨細胞生成。這些發現為OSCAR的膠原識別模式以及合成肽基質因子在破骨細胞生成抑制中的應用提供了有價值的結構見解[36]。
9 結論
骨組織代謝的吸收和形成之間處于微妙動態平衡。骨吸收依賴于破骨細胞相關信號的完美時空協調。破骨細胞在骨量丟失疾病如骨質疏松癥中發揮重要作用,了解和研究這些信號分子對破骨細胞的調節作用,有助于調控破骨細胞的分化和活性,利用這些轉運機制有助于開發新的防治骨質疏松癥等骨代謝疾病的抗骨吸收新療法,以破骨細胞為靶點的藥物,在防治骨量丟失相關疾病方面具有重要意義。
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
