苗藥烏蕨HPLC特征指紋圖譜研究
2021-10-22 01:46:20陳明,段萍
黔南民族醫(yī)專學(xué)報(bào) 2021年3期
關(guān)鍵詞:特征評(píng)價(jià)
陳 明,段 萍
(1.黔南民族醫(yī)學(xué)高等專科學(xué)校,貴州 都勻 558013;2.黔南州食品藥品檢驗(yàn)所,貴州 都勻 558000)
烏蕨(Stenolomachusanum(L.) Ching)為鱗始蕨科烏蕨屬的多年生草本植物,又名烏韭、雉雞尾、大葉金花草、石青葦,分布于長(zhǎng)江流域以南各省份,且蘊(yùn)藏量較大。在民間有“萬(wàn)能解毒藥”之稱,具有確切的療效和悠久的臨床用藥歷史。迄今對(duì)烏蕨化學(xué)成分[1]、藥理作用[2-3]以及質(zhì)量控制方法[4-5]的有關(guān)文獻(xiàn)較多,但特征指紋圖譜的研究未見(jiàn)報(bào)道,為更全面控制烏蕨藥材質(zhì)量,本研究參考相關(guān)文獻(xiàn)[6-7]采用HPLC 法建立了烏蕨藥材的特征指紋圖譜分析方法。
1 儀器與試藥
1.1 儀器 梅特勒AG135、AL204電子天平, SK250LHC超聲波清洗器(上海科導(dǎo)儀器有限公司),島津2401紫外分光光度計(jì),Agilent 1260 DAD VL高效液相色譜儀。處理軟件為“中藥色譜指紋圖譜相似度評(píng)價(jià)系統(tǒng)( 2012-130723版)”(以下簡(jiǎn)稱“相似度評(píng)價(jià)軟件”)。
1.2 試藥 對(duì)照品葒草苷(批號(hào):111777-201302)、牡荊素鼠李糖苷(批號(hào):111668-200602)、牡荊素(批號(hào):111687-201603)、木犀草苷(批號(hào):111720-201106)、原兒茶酸(批號(hào):110809-201205)、原兒茶醛(批號(hào):110810-200205)、香草酸(批號(hào):110776-200402)均購(gòu)于中國(guó)食品藥品檢定研究院;乙腈(天津科密歐化學(xué)試劑有限公司色譜純、分析純),娃哈哈純凈水,其它試劑均為分析純。
收集到不同產(chǎn)地的烏蕨20批(見(jiàn)表1),經(jīng)貴州中醫(yī)藥大學(xué)何順志教授鑒定為鱗始蕨科烏蕨屬Stenolomachusanum(L.) Ching的干燥品。采割地上部分晾干,粉碎,過(guò)3號(hào)篩,置干燥器中保存,備用。
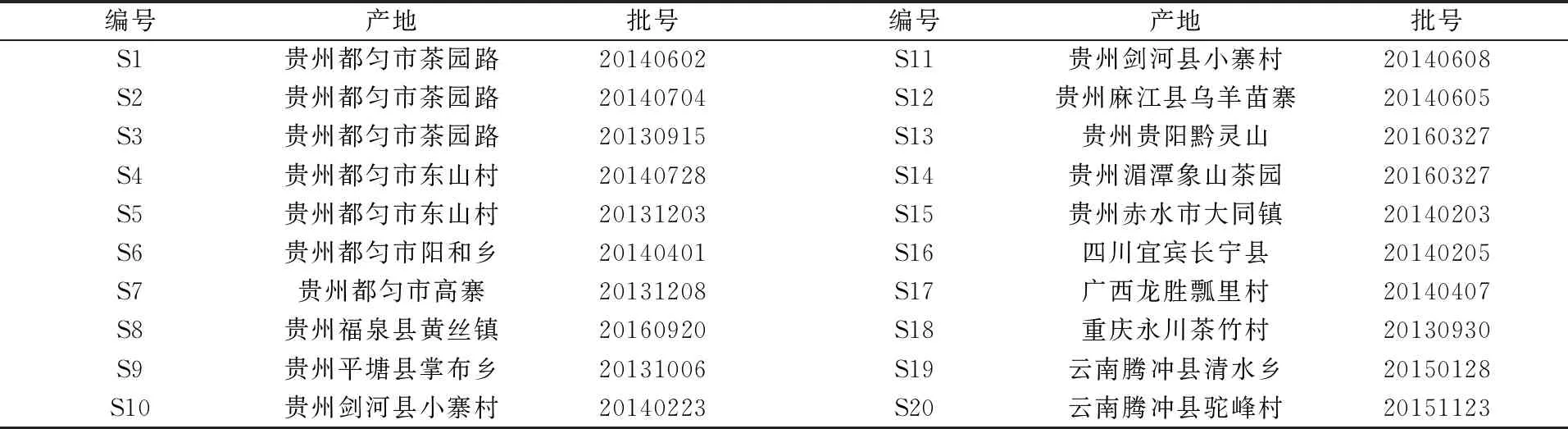
表1 烏蕨樣本表
2 方法與結(jié)果
2.1 混合對(duì)照品溶液的制備 分別取葒草苷、牡荊素、牡荊素鼠李糖苷、木犀草苷、原兒茶醛、原兒茶酸、香草酸適量,精密稱定,加65%乙醇分別制成每1 ml含上述對(duì)照品各1 mg的對(duì)照品儲(chǔ)備液。再精密量取上述各對(duì)照品儲(chǔ)備液適量,加65%乙醇制成每1 ml含葒草苷、牡荊素、牡荊素鼠李糖苷、木犀草苷、原兒茶醛、原兒茶酸、香草酸分別為4.18、17.51、24.84、5.91、10.62、17.00、8.71 μg/ml的對(duì)照品混合溶液。
2.2 供試品溶液的制備 取樣品粉末2.0 g,精密稱定,置于200 ml 具塞錐形瓶中,精密加入65%乙醇50 ml,稱定重量,超聲提取30 min (頻率53 KHz) 后,水浴加熱回流1 h,放冷,再稱定重量,用65%乙醇補(bǔ)足減失的重量,搖勻,0.45 μm微孔濾膜濾過(guò),取續(xù)濾液,作為供試液。
2.3 色譜條件 Agilent Eclipse Plus C18柱 (4.6 mm×250 mm,5 μm);以乙腈(A)-0.4% 磷酸溶液(B)為流動(dòng)相,梯度洗脫(0~10 min,8%→10%A;10~15 min,10%→18%A;15~36 min,18%A;36~37 min,18%→8%A;37~46 min,8%A);檢測(cè)波長(zhǎng)280 nm;柱溫30℃;進(jìn)樣量5 μl;流速1 ml/min。
2.4 方法學(xué)考察
2.4.1 精密度試驗(yàn) 取S1號(hào)供試品溶液,按“2.3”項(xiàng)下色譜條件進(jìn)樣6次,記錄色譜圖,采用 “相似度評(píng)價(jià)軟件”進(jìn)行分析,符合中藥特征色譜的要求。選用葒草苷色譜峰為參照峰,計(jì)算其他共有峰的相對(duì)保留時(shí)間及相對(duì)峰面積的RSD值,共有峰相對(duì)峰面積RSD≤1.2%,相對(duì)保留時(shí)間RSD≤0.5%,表明儀器精密度良好,符合檢測(cè)需求。
2.4.2 重復(fù)性試驗(yàn) 精密稱取S1號(hào)樣品6份,按“2.2”項(xiàng)下方法制備供試液,按“2.3”項(xiàng)下方法檢測(cè),用“相似度評(píng)價(jià)軟件”進(jìn)行分析,6份色譜圖的相似度為1,符合中藥特征色譜的要求。以葒草苷峰為參照峰,計(jì)算各共有峰相對(duì)峰面積、相對(duì)保留時(shí)間的RSD,均小于2.0%,表明方法重復(fù)性良好。
2.4.3 穩(wěn)定性試驗(yàn) 取S1號(hào)供試品溶液,分別于0、5、15、24 h 進(jìn)樣測(cè)定,以葒草苷為參照峰,分別計(jì)算共有峰的相對(duì)保留時(shí)間及相對(duì)峰面積的RSD值。結(jié)果表明,共有峰相對(duì)保留時(shí)間及相對(duì)峰面積RSD 均≤2.0%,表明供試品溶液24 h內(nèi)穩(wěn)定性良好。
2.5 烏蕨HPLC指紋圖譜的建立 取20批烏蕨藥材,按“2.2”項(xiàng)下方法制備供試品溶液,另取“2.1”項(xiàng)下對(duì)照品溶液,按“2.3”項(xiàng)下色譜條件分別進(jìn)樣,記錄HPLC色譜圖。將20批烏蕨的HPLC色譜圖數(shù)據(jù)導(dǎo)入“相似度評(píng)價(jià)軟件”,選擇S1號(hào)樣品圖譜作為參照?qǐng)D譜,進(jìn)行多點(diǎn)校正,色譜峰全峰自動(dòng)匹配,以中位數(shù)法(時(shí)間窗寬度為0.1)生成烏蕨對(duì)照特征圖譜(R)。烏蕨HPLC的對(duì)照品圖譜、對(duì)照特征圖譜及20批烏蕨樣品特征圖譜的匹配圖分別見(jiàn)圖1、2、3。葒草苷的出峰時(shí)間居中,峰面積較大,分離度較好,因此選取該峰作為參照峰(S),計(jì)算20批烏蕨樣品各共有峰的相對(duì)保留時(shí)間,RSD均≤0.6%,表明所得烏蕨指紋圖譜可信度較高。
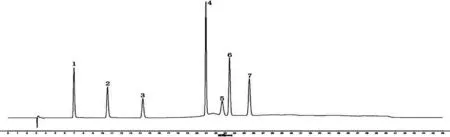
圖1 對(duì)照品HPLC圖譜
2.6 烏蕨藥材相似度評(píng)價(jià) 20批樣品的特征圖譜分析結(jié)果,烏蕨中主要有11個(gè)共有峰,確認(rèn)出6個(gè)色譜峰,分別為原兒茶酸、原兒茶醛、葒草苷、牡荊素鼠李糖苷、牡荊素、木犀草苷,相對(duì)保留時(shí)間分別為0.337、0.503、1.000、1.083、1.217,RSD均≤0.6%。色譜圖數(shù)據(jù)導(dǎo)入“相似度評(píng)價(jià)軟件”,以烏蕨對(duì)照特征圖譜(R)為標(biāo)準(zhǔn),計(jì)算20批樣品與對(duì)照特征色譜圖的相似度,結(jié)果在0.855~0.957之間,表明相似度較好。見(jiàn)表2。
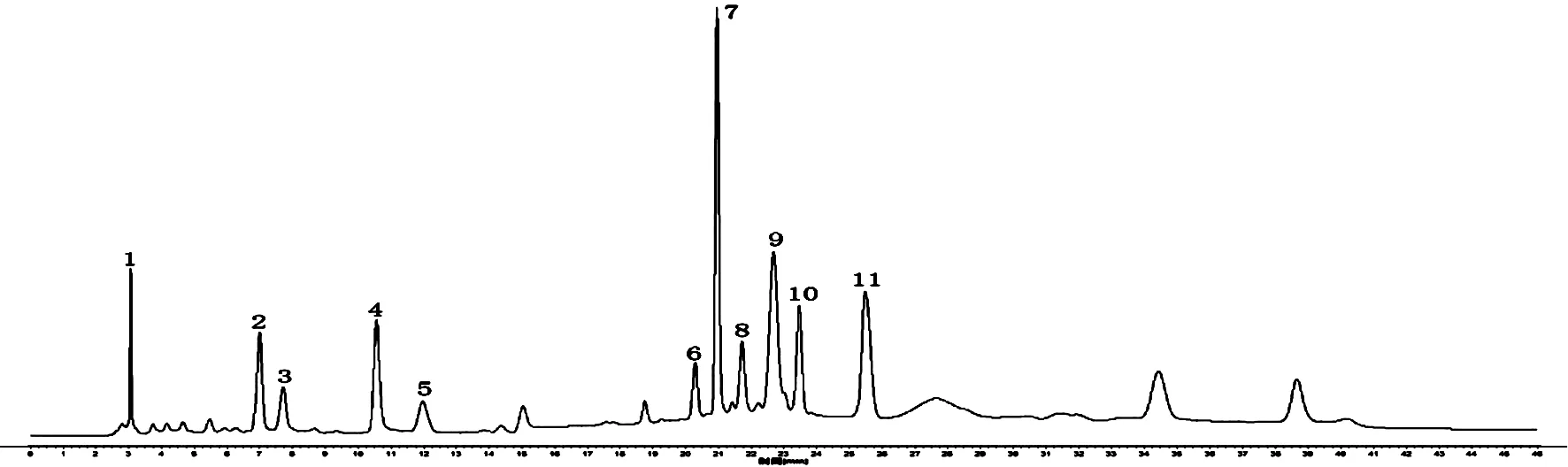
圖2 烏蕨的對(duì)照特征圖譜(R)
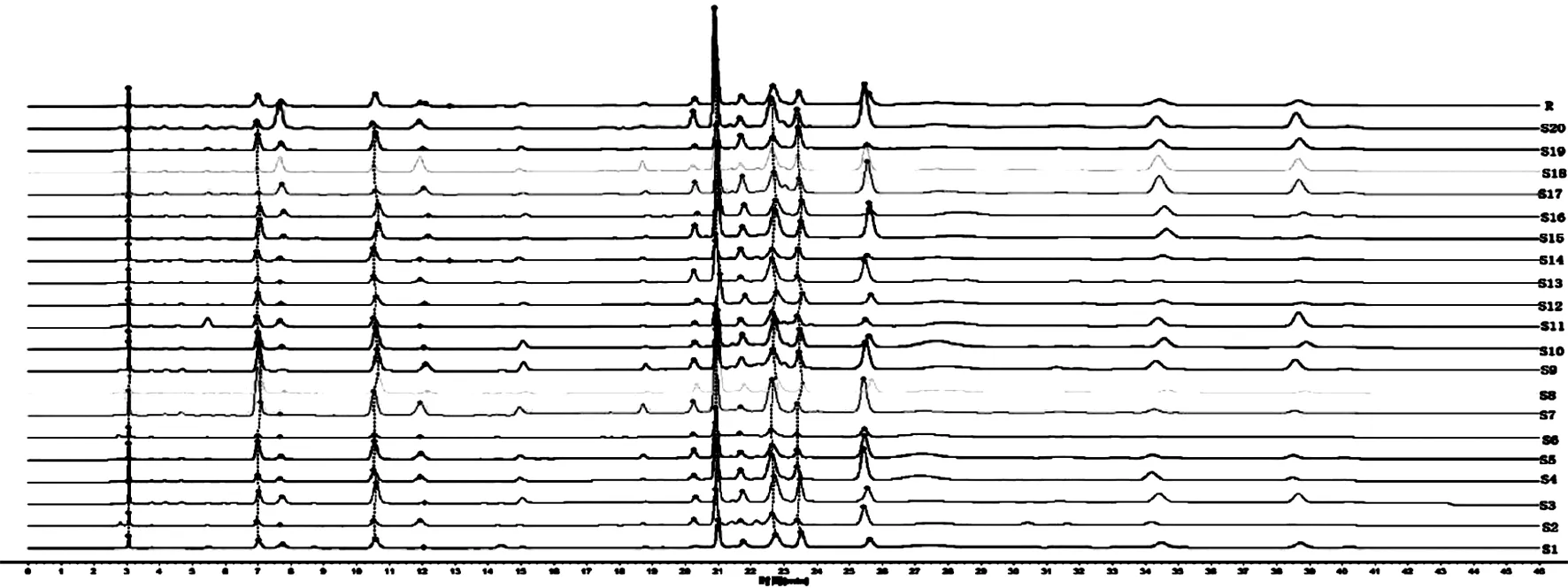
圖3 20批烏蕨的特征圖譜

表2 樣本相似度評(píng)價(jià)
3 討論
3.1 波長(zhǎng)選擇 考察280、254、350 nm波長(zhǎng)下的各色譜峰,280 nm波長(zhǎng)檢測(cè)到的色譜峰信息較全面,且各色譜峰的峰形及分離度較好,因此最終選擇280 nm作為烏蕨HPLC指紋圖譜的檢測(cè)波長(zhǎng)。
3.2 流動(dòng)相選擇 嘗試選擇乙腈-水、甲醇-0.1%醋酸及乙腈-0.4%磷酸3種流動(dòng)相進(jìn)行HPLC 梯度洗脫分析,結(jié)果顯示,乙腈-水、甲醇-0.1%醋酸所得色譜峰較多,但部分峰的峰形、分離度較差,乙腈-0.4%磷酸所得峰形、主要成分分離度較好,故選擇乙腈-0.4%磷酸作為烏蕨HPLC分析測(cè)定的流動(dòng)相。
3.3 云、貴、川、桂、湘采集的20個(gè)不同批次的烏蕨雖有差異,但相似度均≥0.85,可作為烏蕨藥材的質(zhì)量控制方法。
綜上所述,本文采用HPLC特征指紋圖譜分析方法初步建立了苗藥烏蕨質(zhì)量評(píng)價(jià)標(biāo)準(zhǔn),對(duì)有關(guān)機(jī)構(gòu)進(jìn)一步研究開發(fā)苗藥烏蕨有一定參考作用。
猜你喜歡
數(shù)學(xué)小靈通·3-4年級(jí)(2024年2期)2024-05-15 02:02:28
石油瀝青(2021年4期)2021-10-14 08:50:44
世界科學(xué)技術(shù)-中醫(yī)藥現(xiàn)代化(2021年10期)2021-03-02 05:52:06
世界科學(xué)技術(shù)-中醫(yī)藥現(xiàn)代化(2020年2期)2020-07-25 02:05:36
瘋狂英語(yǔ)·新策略(2019年10期)2019-12-13 08:43:28
當(dāng)代陜西(2019年10期)2019-06-03 10:12:04
數(shù)學(xué)小靈通·3-4年級(jí)(2017年9期)2017-10-13 08:10:54
中國(guó)教育技術(shù)裝備(2015年19期)2015-03-01 02:43:07
中國(guó)工程咨詢(2015年2期)2015-02-14 02:59:26
河南科技(2014年23期)2014-02-27 14:19:15
