伴嗜堿性粒細胞增多的兒童急性早幼粒細胞白血病2例并文獻復習
2021-10-28 12:07:34劉俊閃史利歡褚苗孫佳田亮
河南醫學研究 2021年29期
劉俊閃,史利歡,褚苗,孫佳,田亮
(河南省兒童醫院 a.河南省小兒血液醫學重點實驗室;b.血液腫瘤科,河南 鄭州 451161)
急性早幼粒細胞白血病(acute promyelocytic leukemia,APL)是急性髓系白血病(acute myelogenous leukemia,AML)的一種特殊類型,因其特征性基因改變,WHO 2016(修訂版)急性髓系白血病分型將其歸類為AML伴重現性遺傳異常中的APL伴PML-RARA亞型[1]。由于維甲酸的應用,目前 APL被認為是可能被治愈的白血病[2-3]。據文獻報道,兒童APL發生率低,僅占兒童AML的10%[4],伴有嗜堿性粒細胞增多APL尤為少見。本研究報告河南省兒童醫院收治的兩例伴嗜堿性粒細胞增多的APL,并進行文獻復習,以期對該疾病的認識、診斷有所助益。
1 病歷資料
1.1 病例1男,10歲。以面色蒼黃2個月余,皮膚瘀斑1個月余,間斷發熱10 d,咳嗽3 d為代主訴,于2019年9月23日入住河南省兒童醫院。
病史:2個月余前無明顯誘因出現面色蒼黃,無發熱、咳喘、吐瀉,無鞏膜黃染、關節腫痛、皮疹等,家屬未在意,1個月余前無明顯誘因出現皮膚出血點及瘀斑,鼻出血1次,量較大,按壓后可止血,無血尿、血便等,家屬仍未在意;10 d前無明顯誘因出現發熱,熱峰38 ℃,無咳嗽、咳痰、吐瀉等,就診于當地診所,接受口服藥物治療(具體用藥不詳)后體溫正常,3 d前因出現咳嗽、咳痰,伴發熱,熱峰38 ℃,就診于當地醫院,查血常規示血紅蛋白68 g·L-1,血小板21×109L-1,接受甲潑尼龍40 mg、丙種球蛋白2.5 g×6支。
既往史:平素易患“濕疹”。4個月前因“風疹”查血常規示白細胞1.54×109L-1,中性粒細胞0.39×109L-1,血紅蛋白117 g·L-1,血小板158×109L-1,具體用藥不詳。
入院查體:全身皮膚黏膜蒼白,可見散在針尖大小出血點,雙下肢、腰背部可見不規則形狀片狀瘀斑,口唇蒼白,口唇及雙側頰黏膜可見數個綠豆大小血皰,上顎及舌體可見針尖大小出血點。聽診心肺無異常,肝脾肋下未觸及。血常規:白細胞5.77×109L-1,中性粒細胞比率39.8%,淋巴細胞比率30.2%,單核細胞比率29.8%,紅細胞1.98×1012L-1,血紅蛋白69×109L-1,血小板17×109L-1。外周血細胞形態:中性粒細胞百分比8.0%,淋巴細胞百分比44.0%,單核細胞百分比8.0%,原幼細胞40.0%,成熟紅細胞大小形態正常,色素充盈可,血小板散在少見。骨髓形態:骨髓增生活躍,早幼粒比值增高,原粒+早幼粒占72.0%,胞體中等大小,胞漿量較豐富,染藍色,可見多量紫紅色顆粒,少見“柴捆狀”Auer小體;核呈類圓形,可見扭曲,折疊,核染色質細致,核仁顯隱可見,POX染色100%(+),中幼粒及以下階段粒細胞占16.8%。免疫分型:骨髓中約有52.2%的髓系來源異常細胞,表達CD9、CD13、CD33、CD117、CD123,少部分表達CD34;另可見嗜堿性粒細胞約占20.1%,比例增高,考慮急性髓系白血病。46種融合基因篩查:PML-RARa 陽性(Ct=25.74),余項目陰性;PML-RARa 1.48×106copies·mL-1,ABL 3.26×106copies·mL-1,PML-RARa/ABL 45.40%。白血病預后基因篩查:NRAS突變,余項目陰性。FISH:PML-RARa雙融合探針計數200個細胞,發現陽性細胞比率75%;AML1-ETO雙融合探針計數200個細胞,無發現陽性信號,但在計數過程中發現90%的細胞含有四個綠色信號及兩個紅色信號,余項目陰性。染色體:核型47,XY,t(15,17)(q24;q21),+ mar[10];診斷急性早幼粒細胞白血病明確。2019年9月24日開始接受維A酸片口服治療,2019年9月25日接受亞砷酸靜點化療。現規律化療中。
1.2 病例2女,12歲,2019年12月18日患兒無明顯誘因出現陣發性咳嗽,干咳為主,鼻出血1次,量多、不易止血。患兒自訴乏力、懶動。查血常規提示全血細胞減少。門診以“全血細胞減少”收入院。
既往史:平素體質一般,無“肝炎、結核”等傳染性疾病及接觸史,無食物、藥物過敏史,3歲齡因“先天性腸道發育畸形、腸梗阻”接受“腸道切除術”,無外傷史。
入院查體:意識清醒,反應欠佳,貧血貌,全身皮膚蒼黃,無黃染、皮疹、出血點,全身淺表淋巴結未觸及腫大,心肺聽診未聞及明顯異常,肝脾肋下未觸及,胸骨無壓痛。血常規:白細胞1.84×109L-1,紅細胞1.62×1012L-1,血紅蛋白 54 g·L-1,血小板51×109L-1,中性細胞比率29.2%,淋巴細胞比率63.9%,單核細胞比率6.0%,CRP 13.93 mg·L-1。外周血細胞形態:早幼粒細胞2.0%。骨髓形態:骨髓增生極度活躍;異常早幼粒細胞比值增高,原粒+早粒占87.6%,異常早幼粒細胞中等大小,類圓形,胞漿量豐富,染藍色,可見多量顆粒及柴捆狀“Auer”小體(圖1A),可見內外漿,胞核不規則,核染色質細致,核仁顯隱可見,POX染色100%(+)(圖1B),粒系中幼粒及以下階段細胞比值降低,占6.8%,紅系增生受抑制,全片巨核細胞6個,血小板散在少見。免疫分型:骨髓中約有61.7%的髓系來源異常細胞,表達CD9、CD13、CD33、CD64、CD117、CD123、MPO,另可見嗜堿性粒細胞約占18.1%,比率增高,考慮AML(圖2)。白血病篩查基因:PML-RARa陽性(Ct=26.48)。AML預后基因WT1突變(12.46%)。PCR定量:PML-RARa/ABL 50.00%。FISH法查PML/RARa 80%陽性。染色體:47,XX,t(15;17)(q24;q21),+21[12]。頭顱+全脊柱磁共振:(1)顱板和顱底骨質DWI信號增高;(2)脊髓圓錐低位;(3)終絲脂肪附著;(4)片中所示各椎體及其附件、雙側肩胛骨、胸骨、肋骨、髂骨、骶骨T2壓脂信號偏高;(5)雙側胸腔少量積液,右側著。腹部彩超:肝內輕度脂肪沉積。胸部CT:雙肺紋理粗。腹部CT:(1)子宮后下緣偏右點狀高密度影;(2)所示L1兩側橫突細小;(3)脊髓圓錐低位;(4)L3椎體下緣水平至L5椎管內后部可見線狀脂肪密度影,考慮終絲脂肪附著,確診急性早幼粒細胞白血病(低危)。2019年12月19日接受維A酸(25 mg·m-2,共28 d),2019年12月21日接受亞砷酸(0.15 mg·kg-1,共28 d)化療,2020年1月3日接受阿糖胞苷(0.1 g,共5 d)及羥基脲降白細胞,并接受地塞米松針預防誘導分化綜合征。第28天復查骨髓象:骨髓增生減低,原始粒1%。白血病免疫分型(殘留-AML)異常表達細胞比率約5.9%。PCR法查PML-RARa/ABL 50.00%,2020年2月2日接受維A酸(25 mg·m-2,共14 d),亞砷酸(0.15 mg·kg-1,共14 d)化療,并接受二聯鞘注預防中樞神經系統白血病,腦脊液檢查未見異常。現規律化療中。
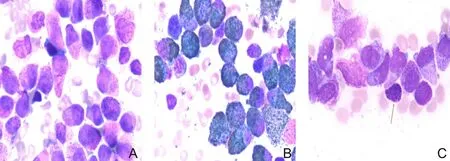
A.APL早幼粒細胞(瑞氏染色,×1 000),箭頭示Auer小體;B.APL早幼粒細胞MPO強陽性(MPO染色,×1 000);C.嗜堿性粒細胞(瑞氏染色,×1 000),箭頭示嗜堿性粒細胞。
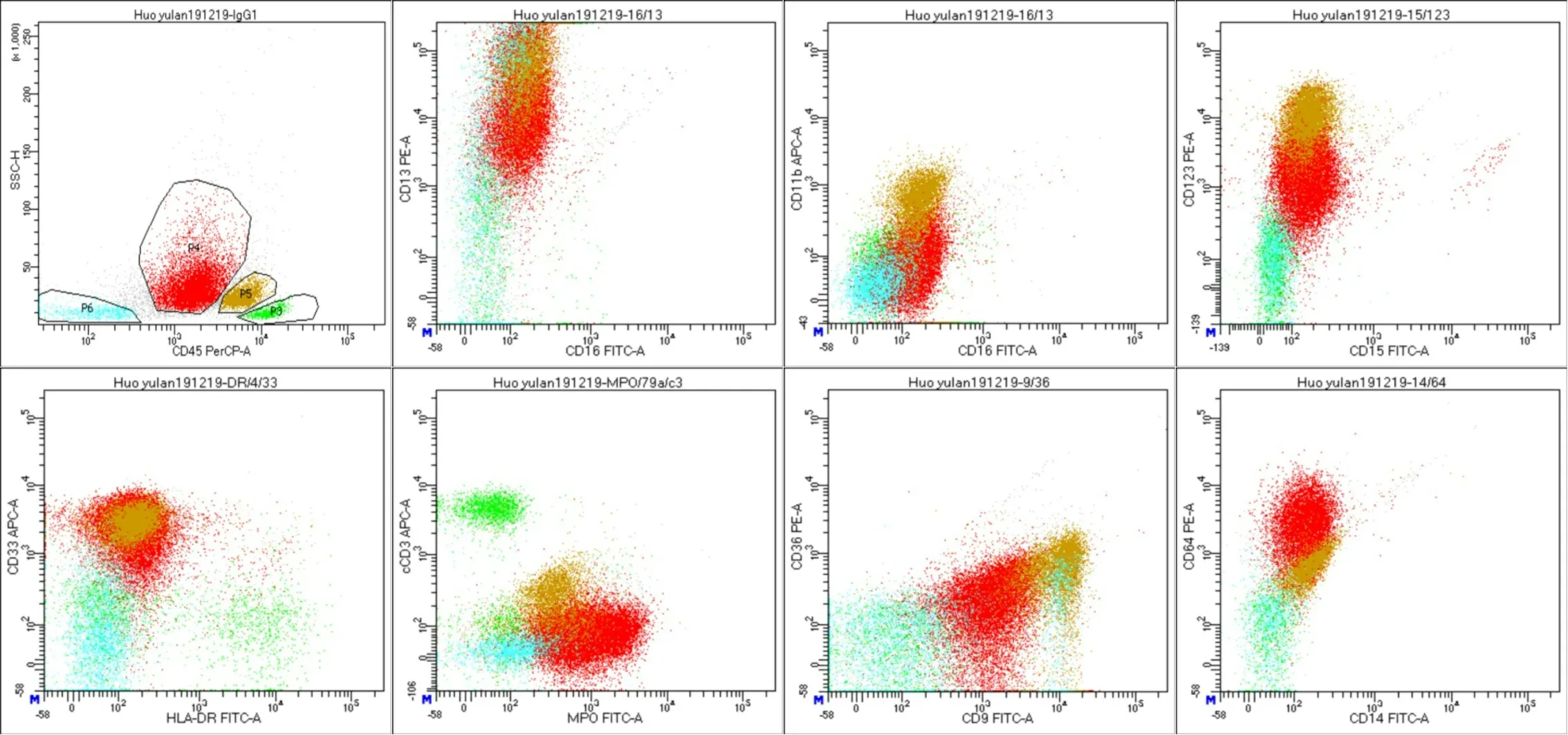
棕黃色為嗜堿性粒細胞;紅色為APL細胞;嗜堿性粒細胞表達CD9、CD13、CD33,CD123,不表達CD117、MPO。
2 討論
急性早幼粒細胞白血病骨髓形態以顆粒增多的異常早幼粒細胞為主(>20%),其胞核大小不一,胞質中有大小不等的嗜苯胺藍顆粒,胞質中常有Auer小體[5-6]。典型APL細胞免疫學表型為CD13、CD33陽性,CD34和HLA-DR陰性,染色體檢查可見t(15;17)(q22;q12)易位,形成PML/RaRa融合基因和變異型[7-8]。 本報道中兩例患者經基因檢測證實,均為PML/RaRa融合基因陽性,流式圖中CD45/SSC設門,發現一群典型的APL細胞,其表型為SSChighCD9+CD13+CD33+CD117+CD123+CD64+CD11b-CD34-HLA-DR-MPO+,與以往文獻中典型APL細胞表型[9-11]不同之處在于,此兩例病例中還發現有一群嗜堿性粒細胞,該群細胞在CD45/SSC散點圖上,位于原始細胞右上方,跟單核細胞位置接近,SSC值偏小,其表型為CD9+CD13+CD33+CD117dim+CD11b+MPO-,弱表達CD22,不表達單核細胞標志CD14、 CD64[12]。有研究報道,異常早幼粒細胞胞質中除嗜天青顆粒外,尚含有大小不一深紫色的嗜堿性顆粒,經甲苯胺藍染色證實,屬于APL的特殊類型,即伴嗜堿顆粒型APL[13]。陳芳等[14]報道APL細胞易伴有嗜堿性顆粒增多,這種伴有嗜堿性顆粒增多的APL細胞SSC稍小,CD45表達較強,表達CD13、CD9和MPO,不表達CD123、CD25、CD22、CD4、CD64和CD14,他們還認為形態學上易將伴有粗大顆粒的APL誤判為嗜堿性粒細胞。但本次報告的2例患兒的嗜堿性粒細胞,雖然形態學嗜堿性粒細胞比率沒有流式比率高,但流式證實其表型為正常嗜堿性粒細胞表型,而并非帶有嗜堿性顆粒的APL細胞。Gotoh等[15]在1988年曾報道過1例成人急性早幼粒細胞白血病同時出現嗜堿性粒細胞的增多,該患者染色體檢查除了有APL典型的t(15;17)染色體基因的異常,同時還有伴有t(9;14)(q34;q22)的易位,所以他們認為此染色體易位是造成嗜堿性粒細胞增多的原因。李佳等[11]也報道過一例成人APL同時出現嗜堿粒細胞增多,其核型正常。而本次報告的兩病例染色體1例+21,1例+mar,均未見到t(9;14)(q34;q22)的易位,可見此易位并非是出現嗜堿性粒細胞增多的原因。不同于之前的報道,本研究中例1患兒AML預后基因可見NRAS突變,例2患兒可見WT1突變,這些基因突變與疾病預后有關[16-20],但未見與嗜堿性粒細胞增多有關的報道。APL伴隨嗜堿性粒細胞比率增高病例比較少見,尤其是兒童病例更是罕見,由于嗜堿性粒細胞的增多與基因異常的關系及對預后的判斷都沒有明確的相關性,而形態學有時又難以將其與含粗大紫黑顆粒的早幼粒細胞區分開,故流式細胞術是檢測嗜堿性粒細胞更敏感更特異的檢測方法。
