鐵死亡與腫瘤免疫的研究進展*
2021-11-01 03:28:38陳麒安劉超群陳怡趙亮
廣東醫學 2021年10期
關鍵詞:信號
陳麒安,劉超群,陳怡,趙亮△
南方醫科大學 1第二臨床醫學院,2基礎醫學院病理學系,3第一臨床醫學院(廣東廣州 510515)
鐵死亡由Stockwell等學者于2012年提出。這是一種鐵依賴性的、由脂質活性氧(reactive oxygen species,ROS)驅動的程序性死亡形式,它在細胞形態、生化、遺傳特征方面顯著不同于凋亡、壞死和自噬等死亡形式。鐵死亡的發生機制為:膜磷脂在非酶或脂氧合酶(lipoxygenase,LOX)等途徑及鐵的催化下形成脂質ROS,脂質ROS持續累積最終導致細胞死亡。細胞對鐵死亡的敏感性主要由谷胱甘肽過氧化物酶4(glutathione peroxidase 4,GPX4)調節。GPX4是一種硒代半胱氨酸酶,它能在還原型谷胱甘肽(glutathione,GSH)存在的條件下將毒性脂質ROS還原為無毒的脂質醇,從而阻止鐵死亡的發生。GSH的合成原料由胱氨酸-谷氨酸逆向轉運體(System Xc-)供應。System Xc-可在將谷氨酸輸出至細胞外的同時,將細胞外的胱氨酸轉運入細胞內,從而促進GSH的合成,維持GPX4的活性。小分子erastin可以通過抑制System Xc-對胱氨酸的轉運,導致GSH耗竭和GPX4失活,從而誘導鐵死亡,而RSL3則通過抑制GPX4的活性直接誘導細胞發生鐵死亡。研究發現,GSH-GPX4對于維持細胞活性尤為重要,在免疫細胞中亦然。此外,GSH耗竭和(或)GPX4失活的腫瘤細胞發生鐵死亡,釋放細胞內源性分子調節免疫反應,同時,免疫系統釋放的細胞因子也可通過調節腫瘤細胞內GSH-GPX4水平,影響細胞對鐵死亡的敏感性(圖1)。
1 GSH-GPX4軸對免疫細胞活性的調節作用
GPX4作為脂質ROS清除的關鍵酶,對維持細胞活性至關重要。事實上,氧化應激在細胞活性的調節中起著雙重作用,它一方面幫助細胞在細胞外環境或細胞內刺激下生存或分化,另一方面卻會引起膜破裂、蛋白質降解、脂質過氧化、DNA損傷、細胞死亡等。在T細胞活化過程中,線粒體ROS對于活化T細胞核因子(nuclear factor of activated T cells,NFAT)、核因子-κB(neclear factor-κB,NF-κB)和激活蛋白-1(activator protein-1,AP-1)等相關轉錄因子的激活至關重要[1]。研究表明,GSH可以通過調節T細胞中ROS依賴的信號通路參與代謝重編程,協調T細胞亞群的分化和能量代謝需求[2],進而影響腫瘤微環境中的免疫調節。此外,鐵死亡誘導劑erastin誘導的脂質過氧化可通過抑制骨形態發生蛋白家族的表達,促進人外周血單核細胞(peripheral blood mononuclear cell,PBMC)向B細胞和自然殺傷細胞的增殖和分化[3]。然而,有研究表明,髓系細胞中GPX4的消耗增加了敗血癥的致死性[4]。GPX4敲除的髓系細胞無法有效清除活性氧,這將增加腸上皮的遺傳不穩定性,進而刺激腸腫瘤的發生及侵襲性生長[5]。另外,GPX4缺失的小鼠T細胞將迅速聚集脂質過氧化物并發生鐵死亡,導致免疫系統無法保護機體免受感染的侵害[6],這提示GPX4在介導T細胞免疫反應中也發揮重要作用。
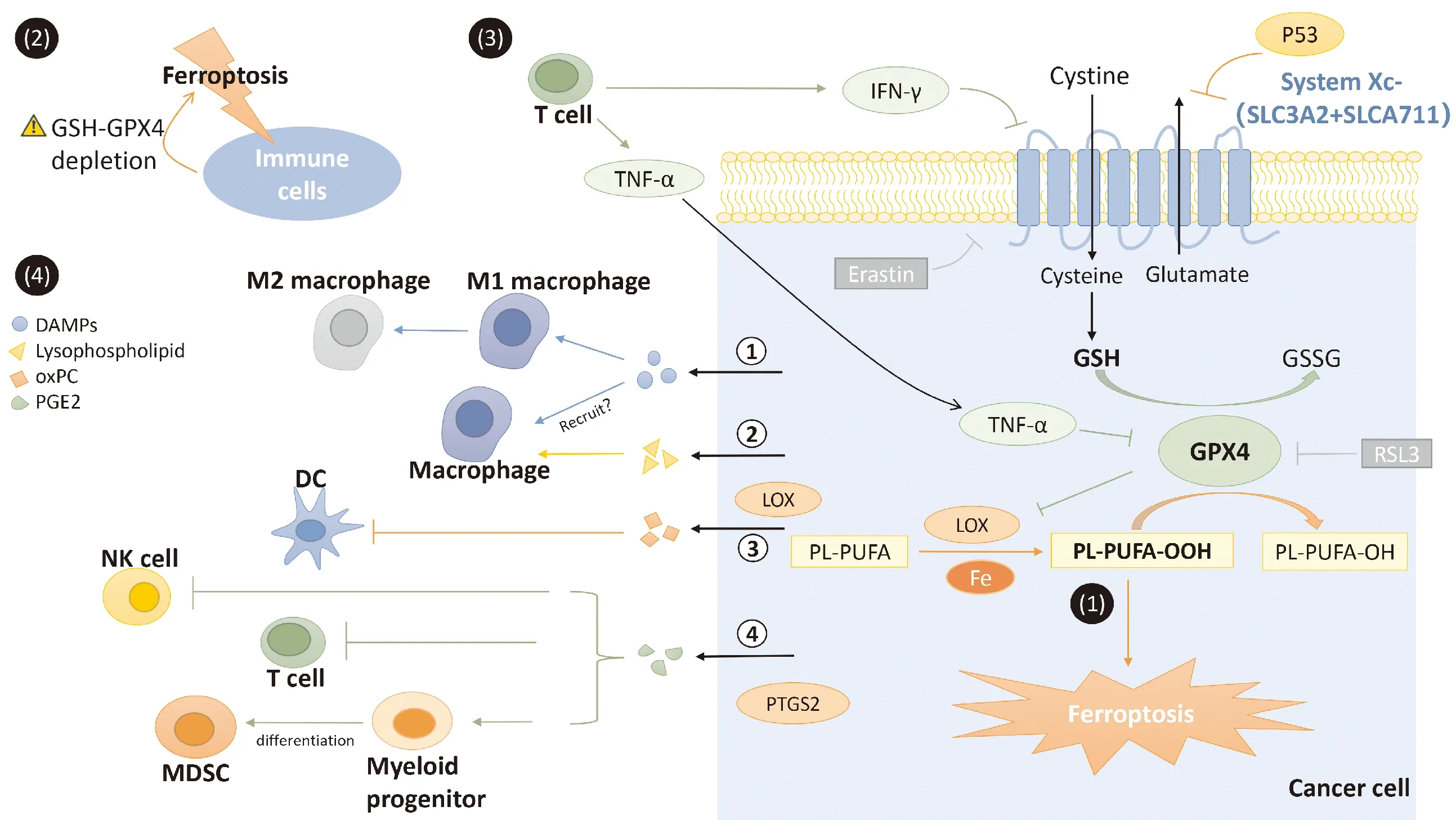
注:(1)鐵死亡發生過程;(2)GSH耗竭和(或)GPX4失活的免疫細胞發生鐵死亡;(3)T細胞釋放IFN-γ、TNF-α調節細胞內GSH-GPX4,從而調節細胞對鐵死亡的敏感性;(4)腫瘤細胞鐵死亡誘發的損傷信號參與調節免疫細胞活性與功能。DAMPs:損傷相關分子模式(damage associated molecular patterns);oxPC:氧化磷脂酰膽堿(oxidized phosphatidylcholine);PGE2:前列腺素E2(prostaglandin E2);樹突狀細胞(dendritic cell);MDSC:髓源性抑制細胞(myeloid-derived suppressor cell)圖1 鐵死亡與腫瘤免疫調節
2 免疫細胞因子對腫瘤細胞GSH-GPX4軸的調節作用
研究發現,CD8+T細胞可通過釋放IFN-γ下調System Xc-的關鍵組成亞單位SLC7A11和SLC3A2的表達,抑制谷氨酸轉運及GSH合成,從而促進腫瘤細胞發生脂質過氧化和鐵死亡。聯合cysteinase(一種可降解胱氨酸和半胱氨酸的工程酶)與免疫檢查點阻滯可能協同地增強T細胞介導的抗腫瘤免疫及誘導細胞鐵死亡的作用[7]。
在黑色素瘤中,免疫活性升高引起的炎癥性微環境可促進黑色素瘤細胞的去分化,這種去分化狀態是黑色素瘤細胞對靶向治療和免疫治療交叉耐藥的根源。實驗證實,用TNF-α和IFN-γ刺激黑色素瘤細胞可分別導致NF-κB或STAT1信號通路的激活,誘導細胞去分化,提高其對erastin和RSL3誘導的鐵死亡的敏感性。其機制可能與去分化黑色素瘤細胞中GSH的水平相關[8]。因此,免疫細胞因子通過誘導黑色素瘤細胞對鐵死亡的敏感性,可以清除去分化狀態的細胞群,進一步阻止具有免疫抑制能力的黑色素瘤細胞累積。可見,這種聯合治療可以在有效殺死腫瘤細胞的同時避免了腫瘤細胞的免疫逃逸。
3 腫瘤細胞鐵死亡誘發的損傷信號參與調節免疫細胞活性與功能
研究表明,凋亡細胞可以通過釋放“找我(find me)”信號吸引巨噬細胞,同時,凋亡細胞表面暴露的“吃我(eat me)”信號則誘導巨噬細胞將其吞噬[9]。可以想象,發生鐵死亡的腫瘤細胞,也釋放類似的信號發揮免疫調節作用。在體外,巨噬細胞有效吞噬了發生鐵死亡的腫瘤細胞,支持了此類信號的存在[10]。
3.1 腫瘤細胞鐵死亡釋放損傷相關分子模式 通過壞死性途徑死亡的細胞(如壞死、焦亡),其特征是細胞膜破裂并釋放細胞內成分,如損傷相關分子模式(damage associated molecular patterns,DAMPs),以吸引巨噬細胞,提高抗原提呈效率。DAMPs包含“find me”和“eat me”信號,如鈣網蛋白、ATP、HMGB1等[11]。實驗證明,發生鐵死亡的腫瘤細胞可通過自噬依賴性方式釋放高遷移率族蛋白1(high mobility group box 1,HMGB1)[12]。HMGB1是一種細胞核內的非組蛋白,它被釋放到胞外后,作為DAMPs,通過與RAGE、TLR2、TLR4等受體結合,激活相關信號通路,從而誘導免疫細胞的活化及細胞因子的產生[13]。在心臟缺血/再灌注損傷模型中,鐵死亡細胞通過釋放DAMPs,激活固有免疫,募集嗜中性粒細胞[14]。在胰腺導管腺癌中,發生自噬依賴性鐵死亡的腫瘤細胞可以釋放Kras作為一種DAMPs,通過RAGE途徑誘導巨噬細胞脂肪酸氧化,進而向M2型極化,誘導免疫抑制[15]。
3.2 腫瘤細胞鐵死亡釋放脂質介質 對發生鐵死亡的細胞進行脂質分析,發現有大量雙氧合和三氧合的含磷脂酰乙醇胺(phosphatidylethanolamine,PE)的多不飽和脂肪酸(polyunsaturated fatty acid,PUFA)積累,這可能是由15-LOX催化產生的[16]。雖然細胞膜外氧化的PE及其氧化分解產物、水解產物的生物學作用尚未可知,但已證實在凋亡細胞中,溶血磷脂可作為免疫調節信號吸引抗原提呈細胞[17]。另外,有實驗發現,15-LOX產生的脂質介質可調節樹突狀細胞(dendritic cell,DC)的成熟并調節適應性免疫反應,如氧化磷脂酰膽堿(phosphatidylcholine,PC)可通過激活轉錄因子NRF2抑制DC成熟,并抑制輔助性T細胞17(helper T cell,Th17)細胞的分化[18]。然而,在鐵死亡環境中釋放的脂質介質是否也發揮著同樣的作用還有待進一步的研究證實。
研究發現,發生鐵死亡的腫瘤細胞中,除了LOX產物增加外,還有PGE2的大量釋放,這可能與前列腺素內過氧化物合酶2(prostaglandin-endoperoxide synthase 2,PTGS2)的表達顯著上調有關[19]。目前已經證實,PGE2作為促炎因子發揮作用的同時,也具有免疫抑制活性。PGE2誘導的免疫抑制作用如下:(1)抑制NK細胞的活性及其分泌CCL5和XCL1的能力,從而抑制腫瘤微環境中DC的招募[20];(2)促進DC分泌的細胞因子從Th1 型向 Th2 型轉換,從而降低CD8+T細胞的活性[21];(3)誘導髓樣祖細胞分化為髓源性抑制細胞(myeloid-derived suppressor cell,MDSC),增強MDSC介導的免疫抑制及其誘導產生Treg細胞的能力,進而促進腫瘤進展[22]。
綜上所述,鐵死亡細胞釋放的脂質介質可能在免疫系統激活的多個步驟中發揮調節作用,包括抗原加工、提呈、免疫細胞的成熟、遷移。但在不同條件下,鐵死亡釋放的不同信號有可能發揮著促進免疫或抑制免疫兩種截然相反的作用。
3.3 GPX4在鐵死亡與免疫調節中的作用 細胞內促炎脂質介質的生物合成多由LOX和PTGS催化而來,這些酶的活性則受GSH-GPX4的調控[23]。因此,GSH耗竭和(或)GPX4失活的腫瘤細胞內LOX的酶活性升高,脂質ROS及促炎性細胞因子的合成增多。這些細胞在發生鐵死亡的同時,可以釋放大量細胞內源性分子(包括DAMPs、脂質介質等)激活免疫系統,誘導中性粒細胞和巨噬細胞分泌促炎因子,如TNF-α、IL-1β等[24]。TNF-α和IL-1β可以誘導PTGS2活化,此外TNF-α還可使GPX4持續下調[25],這些效應最終都導致LOX、PTGS2的酶活性升高,進一步促進了鐵死亡及免疫炎癥反應。研究表明,固有免疫系統的細胞通過分泌白三烯、hepoxilins等,與PTGS和LOX酶聯合維持炎癥狀態,并且最終通過分泌resolvins和lipoxin使炎癥消退[24]。這種自發放大循環(auto-amplification loop)也發生在其他非凋亡性細胞死亡形式中,并且最終可導致組織損傷和器官衰竭[26]。可以想象,如果GPX4水平持續下降,固有免疫系統沒有及時釋放消除炎癥的信號,可能會導致持續的慢性炎癥,引起腫瘤環境中的免疫抑制以及免疫炎癥性的病理損傷(如缺血/再灌注損傷)等。
4 腫瘤治療中的鐵死亡與免疫調節
腫瘤免疫治療是一種通過調節人體的免疫防御機制殺傷腫瘤細胞的治療方法,因其不良反應小、特異性強等優點而備受關注。近年來,不斷有用于免疫治療的新靶點和新技術出現。其中,重組腺病毒P53注射液作為世界上第1個基因治療產品,已被應用于多種惡性腫瘤的治療。野生型p53是一種抑癌基因,它除了能夠導致細胞周期阻滯、促進細胞凋亡以外,還與抗腫瘤免疫密切相關[27]。研究發現,在腫瘤細胞中,P53蛋白大量積聚,并作為腫瘤相關抗原誘發機體免疫反應,發揮抗瘤效應[28]。目前已有許多研究表明,P53在腫瘤細胞鐵死亡的調節中發揮重要作用。在人骨肉瘤與人乳腺腺癌細胞中,P53可以通過與SLC7A11基因的啟動子區域結合,抑制SLC7A11的轉錄,進而導致細胞內GSH合成減少,對erastin誘導的鐵死亡敏感性增加[29]。因此,我們設想P53誘導的免疫治療是否可以與鐵死亡誘導劑相結合,提高抗腫瘤治療的療效。
如何有效地將免疫檢查點療法與傳統的腫瘤治療方案相結合是目前腫瘤治療面臨的一個巨大挑戰。有研究發現,放射治療誘導的共濟失調毛細血管擴張突變基因(ataxia telangiectasia-mutated gene,ATM)可以協同免疫治療活化的CD8+T細胞,抑制GSH的合成,從而促進腫瘤細胞脂質過氧化物積累,誘導鐵死亡[30]。該研究提示鐵死亡激動劑可以作為一種放射治療及免疫治療的增敏劑,在兩者聯合治療中起橋梁作用。光動力療法(photodynamic therapy,PDT)是一種新型的腫瘤治療方法,目前已有許多研究發現PDT對于腫瘤微環境中的免疫調節有重要作用,PDT與免疫的聯合治療也備受關注。據報道,復合納米藥物SRF@Hb-Ce6[由鐵死亡誘導劑索拉菲尼(sorafenib,SRF)與血紅蛋白(hemoglobin,Hb)、光敏劑chlorin e6(Ce6)組成的復合納米藥物)],可以將鐵死亡與PDT相結合,具有良好的協同抗腫瘤活性。實驗證明,SRF@Hb-Ce6通過募集腫瘤組織中的CD8+T細胞,增加IFN-γ分泌,協同SRF促進腫瘤細胞發生鐵死亡[31]。由此可見,明確免疫與鐵死亡的相互調節機制,對于將鐵死亡與免疫治療及其他抗腫瘤治療方法聯合,以提高抗腫瘤療效具有重要意義。
5 展望
鐵死亡的發現,為許多疾病的發生機制闡明提供依據,也為腫瘤治療開辟了新途徑。鐵死亡的發生與細胞內鐵、脂質ROS、谷胱甘肽的代謝密切相關,許多分子(如ACSL4、LOX、NADPH、CoQ10)則可通過調節以上三要素調控細胞對鐵死亡的敏感性。目前不斷有研究發現新的鐵死亡調控分子機制及可能的抗腫瘤靶點,然而,關于鐵死亡背景下的壞死性炎癥及免疫調節仍存在一些問題:(1)除了GSH-GPX4軸以外,參與鐵死亡的鐵離子是否也受到免疫因子的調控;(2)LOX、PTGS等不同酶的催化產物是否會引起不同的免疫應答;(3)腫瘤細胞發生的鐵死亡除了殺傷腫瘤細胞外,是否誘導抗腫瘤免疫或者抑制免疫應答。進一步明確這些問題,對于闡明疾病的發生機制,指導如何聯合鐵死亡與免疫用于腫瘤等疾病的治療具有重要臨床意義。
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
