制備方法對(duì)Al2O3-CeO2物化性質(zhì)及CO2加氫制甲醇催化性能的影響
2021-11-03 11:51:28范興其姚夢(mèng)琴王曉丹曹建新
人工晶體學(xué)報(bào) 2021年9期
范興其,姚夢(mèng)琴,劉 飛,王曉丹,曹建新
(1.貴州大學(xué)化學(xué)與化工學(xué)院,貴陽(yáng) 550025; 2.貴州大學(xué),貴州省綠色化工與清潔能源技術(shù)重點(diǎn)實(shí)驗(yàn)室,貴陽(yáng) 550025;3.貴州大學(xué),貴州省工業(yè)廢棄物高效利用工程研究中心,貴陽(yáng) 550025)
0 引 言
自工業(yè)革命以來(lái),頻繁的人類活動(dòng)和不可再生化石能源的使用造成了大量CO2溫室氣體的排放,其資源化利用已迫在眉睫[1-2]。近年來(lái),隨著綠色能源在制氫領(lǐng)域的快速發(fā)展,將高能氫分子(H2)在熱催化條件下還原CO2轉(zhuǎn)化為更高價(jià)值的化學(xué)品、基礎(chǔ)化工原料及燃料等具有重要意義[3-5]。甲醇是一種重要的基礎(chǔ)化工原料,也可以作為燃料使用,同時(shí)還是一種潛在的能源載體[6-8]。因此,CO2加氫制甲醇反應(yīng)路線備受關(guān)注。
盡管合成氣制甲醇工藝已實(shí)現(xiàn)工業(yè)化生產(chǎn),但受制于CO2分子的熱力學(xué)穩(wěn)定性,高性能催化劑的設(shè)計(jì)開(kāi)發(fā)仍是CO2加氫制甲醇工藝面臨的巨大挑戰(zhàn)之一[9-11]。圍繞CO2加氫制甲醇路線,相繼報(bào)道了金屬[12-13]、金屬氧化物[14-15]及金屬間化合物[16-17]催化體系。金屬基催化劑因易于燒結(jié)而適用于低溫合成工藝,而復(fù)合氧化物因具有更高熱穩(wěn)定性而適宜于更高的反應(yīng)溫度[18-19]。已有報(bào)道相繼開(kāi)發(fā)出ZnO-ZrO2、In2O3-ZrO2、Ga2O3-ZrO2、Cd2O3-ZrO2、ZnO-Al2O3及ZnO-Ga2O3等復(fù)合氧化物[20-25]。Wang等[20]報(bào)道了ZnO-ZrO2復(fù)合氧化物催化CO2加氫制甲醇,在320 ℃和5 MPa條件下,CO2轉(zhuǎn)化率為10%,甲醇選擇性為80%,表現(xiàn)出較好的抗燒結(jié)性能和抑制逆水煤氣變換反應(yīng)的作用。Yang等[21]報(bào)道了In2O3/m-ZrO2復(fù)合氧化物催化CO2加氫制甲醇,在320 ℃和3 MPa條件下,CO2轉(zhuǎn)化率為11%,甲醇選擇性為78%。Feng等[22]報(bào)道了GaZrOx復(fù)合氧化物催化CO2加氫制甲醇,在330 ℃和3 MPa條件下,CO2轉(zhuǎn)化率為9%,甲醇選擇性為73%。研究發(fā)現(xiàn)[26-27],復(fù)合氧化物晶粒大小、孔結(jié)構(gòu)、比表面積及氧空位缺陷等與催化性能密切相關(guān)。而復(fù)合氧化物物化性質(zhì)調(diào)變又與制備方法密切相關(guān),精準(zhǔn)調(diào)控兩相界面化學(xué)、物理和幾何性質(zhì)是實(shí)現(xiàn)最佳協(xié)同作用的關(guān)鍵。另外,微流控連續(xù)共沉淀法因具有獨(dú)特的微米級(jí)通道優(yōu)勢(shì)可對(duì)合成過(guò)程的傳熱和傳質(zhì)進(jìn)行強(qiáng)化,有利于納米晶粒的調(diào)控生長(zhǎng)和物化性質(zhì)的調(diào)變[28-30]。You等[29]采用微流控連續(xù)共沉淀法制備了晶粒尺寸為44~100 nm、產(chǎn)率為99%的球形γ-CuI納米晶粒,明顯優(yōu)于共沉淀工藝。另外,Wang等[30]采用微流控連續(xù)共沉淀法合成了ZnO-ZrO2復(fù)合氧化物,該催化劑對(duì)CO2加氫制甲醇反應(yīng)表現(xiàn)出優(yōu)異催化性能,在反應(yīng)溫度320 ℃和反應(yīng)壓力3 MPa條件下,CO2轉(zhuǎn)化率為9.2%,甲醇選擇性達(dá)到93.1%。
據(jù)此,為進(jìn)一步探究復(fù)合氧化物復(fù)合界面性質(zhì)對(duì)CO2加氫制甲醇反應(yīng)的影響,建立復(fù)合氧化物的構(gòu)效關(guān)系,鑒于微流控技術(shù)獨(dú)有的高混合效率和高傳質(zhì)傳熱特性,在課題組對(duì)雙金屬氧化物組分篩選的基礎(chǔ)上,本文提出采用微篩孔型微反應(yīng)器制備Al2O3-CeO2復(fù)合氧化物,并以液相共沉淀法、浸漬法和物理共混法為參考,探究了更有利于CO2加氫制甲醇反應(yīng)的復(fù)合界面性質(zhì)。與現(xiàn)有其他復(fù)合氧化物催化體系比較,微流控連續(xù)共沉淀法制得了尺度更為均一,且氧空位更為豐富的納米Al2O3-CeO2雙金屬?gòu)?fù)合氧化物,在CO2加氫制甲醇工藝中表現(xiàn)出優(yōu)異的催化性能。
1 實(shí) 驗(yàn)
1.1 催化劑制備
1.1.1 微流控連續(xù)共沉淀法
采用微流控連續(xù)共沉淀法制備Al2O3-CeO2復(fù)合氧化物。微流控連續(xù)共沉淀工藝示意圖如圖1所示,微反應(yīng)器的尺寸為6 cm×5.5 cm×3 cm,微篩孔為0.22 mm。微反應(yīng)器由兩個(gè)不銹鋼板和不銹鋼濾膜組成,并由不銹鋼濾膜把微反應(yīng)器分為分散相通道和連續(xù)相通道。按Al/(Al+Ce)摩爾比1∶5,將3.4 g的Al(NO3)3·9H2O和20.2 g的Ce(NH4)2(NO3)6溶于去離子水配制成0.2 mol/L的混合溶液作為分散相,4.6 g的(NH4)2CO3溶于去離子水中形成0.2 mol/L的溶液作為連續(xù)相。微反應(yīng)合成體系在75 ℃條件下完成,分別采用平流泵將上述分散相和連續(xù)相注入微反應(yīng)器中進(jìn)行反應(yīng),兩相流量均設(shè)為25 mL/min。反應(yīng)前驅(qū)體于75 ℃下老化2 h,冷卻至室溫,分別用去離子水和無(wú)水乙醇各洗滌3次,80 ℃干燥6 h,500 ℃焙燒3 h制得Al2O3-CeO2復(fù)合氧化物,記為Al2O3-CeO2-MR。
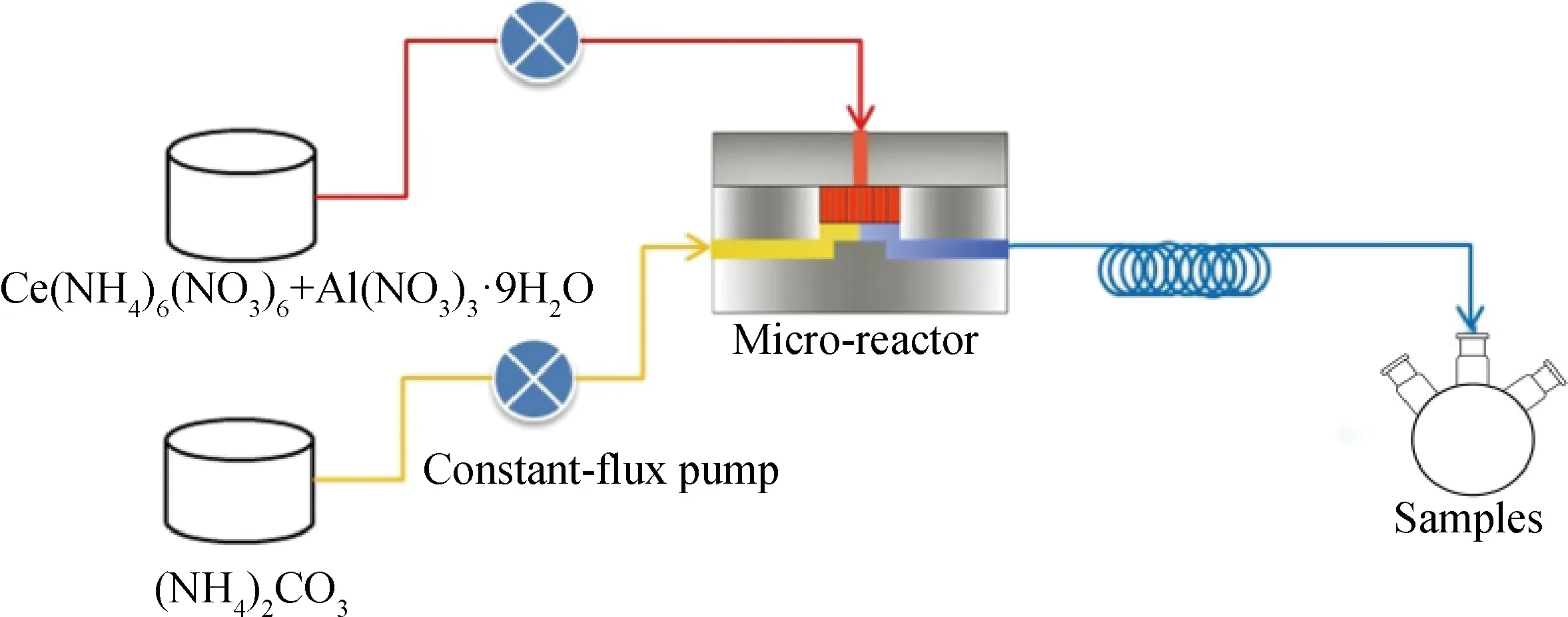
圖1 微流控連續(xù)共沉淀工藝示意圖Fig.1 Schematic diagram of microfluidic continuous co-precipitation process
單一Al2O3、CeO2催化劑的制備是將上述制備工藝中的分散相分別替換為單一的Al(NO3)3·9H2O與Ce(NH4)2(NO3)6溶液。
TiO2-CeO2和La2O3-CeO2復(fù)合氧化物的制備是按相同的摩爾比(1∶5),將分散相體系中的Al(NO3)3·9H2O分別替換為T(mén)iCl4與La(NO3)3·6H2O溶液。
1.1.2 傳統(tǒng)共沉淀法
采用傳統(tǒng)共沉淀法制備Al2O3-CeO2復(fù)合氧化物。按Al/(Al+Ce)摩爾比1∶5,將3.4 g的Al(NO3)3·9H2O和20.2 g的Ce(NH4)2(NO3)6溶于去離子水配制成0.2 mol/L的混合溶液,4.6 g的(NH4)2CO3溶于去離子水配制成0.2 mol/L的沉淀劑溶液。將上述沉淀劑以5 mL/min速度逐滴加入到混合溶液中,前驅(qū)體于75 ℃下老化2 h,冷卻至室溫,分別用去離子水和無(wú)水乙醇各洗滌3次,80 ℃干燥6 h,500 ℃焙燒3 h制得Al2O3-CeO2復(fù)合氧化物,記為 Al2O3-CeO2-CP。
1.1.3 浸漬法
采用等體積浸漬法制備Al2O3-CeO2復(fù)合氧化物。按Al/(Al+Ce)摩爾比1∶5,配制濃度為6 mol/L的Al(NO3)3·9H2O溶液,再稱取2 g上述微流控連續(xù)共沉淀法制備的CeO2催化劑,常溫下量取5 mL的Al(NO3)3·9H2O溶液逐滴滴加到CeO2催化劑中,浸漬24 h后,在80 ℃下干燥6 h,500 ℃下焙燒3 h,制得Al2O3-CeO2復(fù)合氧化物,記為Al2O3-CeO2-IM。
1.1.4 物理共混法
按Al/(Al+Ce)摩爾比1∶5,稱取0.5 g的Al2O3和6.2 g的CeO2進(jìn)行充分混合,制得物理共混樣品記為Al2O3-CeO2-PM。
1.2 催化劑表征
采用Bruker D8衍射儀表征催化劑的晶相結(jié)構(gòu),在40 kV電壓、40 mA電流及Cu Kα輻射源(λ=0.154 178 nm)的條件下進(jìn)行測(cè)試,掃描范圍為5°~90°,步長(zhǎng)為0.02°。采用Thermo Fisher K-Alpha光電子能譜(XPS)測(cè)定催化劑表面元素的分布和氧空位濃度。在加速電壓為30 kV的ZEISS ΣIGMA掃描電鏡(SEM)和加速電壓為200 kV的Phillips FEI Talos-S高分辨透射電鏡(TEM)上觀察催化劑的微觀結(jié)構(gòu)。用Micromeritics ASAP 2020 M分析儀表征催化劑的比表面積和孔結(jié)構(gòu)。使用Micromeritics Auto Chem 2920化學(xué)吸附分析儀進(jìn)行程序升溫吸附測(cè)試,在CO2/NH3-TPD測(cè)試中,樣品在300 ℃下的He氣中熱處理1 h,再經(jīng)CO2或NH3吸附60 min,以10 ℃/min的速率勻速升溫到700 ℃,TCD對(duì)脫附的CO2或NH3進(jìn)行檢測(cè)。用傅里葉變換紅外光譜儀(Thermo Fisher Nicolet iS50)進(jìn)行原位漫反射傅里葉變換紅外光譜研究,在100 mL/min、320 ℃的N2氣流中,吹掃樣品1 h,在320 ℃和1 MPa下,通入原料混合氣(V(H2)∶V(CO2)∶V(N2)=72∶24∶4),以8 cm-1的分辨率掃描64次,收集圖譜。
1.3 催化劑評(píng)價(jià)
催化劑性能評(píng)價(jià)在加壓固定床反應(yīng)器上進(jìn)行,取0.5 g的催化劑裝填在固定床不銹鋼反應(yīng)管的恒溫區(qū)(反應(yīng)管上下部分用石英棉裝填),在儀器檢漏合格后,通入反應(yīng)原料氣(V(H2)∶V(CO2)∶V(N2)= 72∶24∶4),反應(yīng)壓力為3 MPa,反應(yīng)溫度為320 ℃,體積空速為9 000 mL·g-1·h-1。采用GC7820型氣相色譜儀(TCD)在線檢測(cè)反應(yīng)產(chǎn)物中CO、CO2和H2的含量,經(jīng)過(guò)Plot-Q毛細(xì)管色譜柱分離的氣體使用氫火焰離子化檢測(cè)器(FID)檢測(cè)甲烷、甲醇及二甲醚含量。采用C元素歸一法,按下列公式計(jì)算CO2轉(zhuǎn)化率及CO、甲醇、二甲醚、甲烷選擇性。
(1)
(2)
(3)
(4)
(5)
式中:Ax為不同組分的色譜峰面積(其中:ACH4-FID、ACH3OH、ACH3OCH3由FID檢測(cè)器測(cè)得,ACO、ACO2由TCD檢測(cè)器測(cè)得);fCH4/CH3OH/CH3OCH3為甲烷、甲醇、二甲醚相對(duì)于甲烷的相對(duì)校正因子;fCO/CO2為CO、CO2相對(duì)于甲烷的相對(duì)校正因子
(6)
(7)
式中:ACH4-TCD為T(mén)CD圖譜中甲烷的色譜峰面積;ACH4-FID/CH3OH/CH3OCH3為FID圖譜中甲烷、甲醇、二甲醚的色譜峰面積;fCH4-TCD/fCH4-FID=4/1(fCH4-FID值為1);Mx為不同組分的摩爾質(zhì)量(g/mol)。
2 結(jié)果與討論
2.1 Al2O3-CeO2復(fù)合氧化物的物化性質(zhì)
不同方法制得Al2O3-CeO2復(fù)合氧化物的XRD圖譜及對(duì)應(yīng)的晶格應(yīng)力曲線如圖2所示。由圖2(a)可知,合成的單一CeO2樣品與PDF No. 65-297(2θ=28.5°、33.1°、47.4°、56.3°、59.1°)完全一致,屬立方晶相結(jié)構(gòu)c-CeO2。Al2O3-CeO2-PM物混樣品僅出現(xiàn)CeO2特征衍射峰,這與Al2O3為無(wú)定型結(jié)構(gòu)有關(guān),且從衍射峰強(qiáng)度來(lái)看,兩相物理混合時(shí),無(wú)定型的Al2O3對(duì)CeO2結(jié)晶度影響較小。然而,與Al2O3-CeO2-PM物混樣品類似,Al2O3-CeO2-IM、Al2O3-CeO2-CP及Al2O3-CeO2-MR樣品也僅有立方晶相結(jié)構(gòu)CeO2特征衍射峰,不同的是,這些衍射峰強(qiáng)度減弱,且出現(xiàn)明顯寬化現(xiàn)象,這可能與采用的浸漬和共沉淀制備工藝有關(guān)。對(duì)比發(fā)現(xiàn),CeO2晶相對(duì)應(yīng)的(111)、(200)和(220)晶面衍射角向高角度偏移,根據(jù)布拉格定律,這可能是較小離子半徑的Al3+(0.051 nm)進(jìn)入較大離子半徑的Ce4+(0.087 nm)的晶格中,使部分晶面間距減小所致[31]。
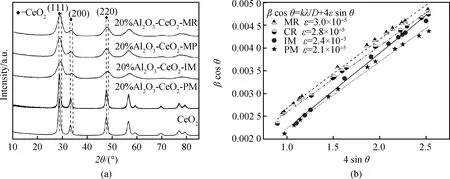
圖2 (a)不同Al2O3-CeO2復(fù)合氧化物的XRD圖譜;(b)不同Al2O3-CeO2復(fù)合氧化物的晶格應(yīng)力曲線Fig.2 (a) XRD patterns of various Al2O3-CeO2 composite oxides; (b) lattice stress curves of various Al2O3-CeO2 composite oxides
圖2(b)是經(jīng)Williamson-Hall方程[32]計(jì)算得到的不同樣品的晶格應(yīng)力曲線,曲線斜率的大小代表Al2O3/CeO2組分間相互作用的強(qiáng)弱。由圖可知,與物混樣品Al2O3-CeO2-PM(2.1×10-5)相比,浸漬樣品Al2O3-CeO2-IM(2.4×10-5)形成的曲線斜率有所提高,但更大的曲線斜率出現(xiàn)在共沉淀樣品中。與傳統(tǒng)共沉淀樣品Al2O3-CeO2-CP(2.8×10-5)相比,微流控連續(xù)共沉淀法制得的Al2O3-CeO2-MR(3.0×10-5)樣品曲線斜率最大,表明該樣品中Al2O3/CeO2組分間相互作用最強(qiáng),預(yù)示著大量晶格缺陷的形成。
Al2O3-CeO2復(fù)合氧化物的XPS圖譜如圖3所示。不同樣品中Ce結(jié)合能的變化,可以直接反應(yīng)Al和Ce界面的相互作用[33]。由Ce 3d軌道圖譜(見(jiàn)圖3(a))可知,CeO2在v4和u4的結(jié)合能分別為898.5 eV與916.4 eV,Al2O3-CeO2-CP(v4=916.58 eV、u4=898.56 eV)和Al2O3-CeO2-MR(v4=916.61 eV、u4=898.6 eV)結(jié)合能較Al2O3-CeO2-IM(v4=916.52 eV、u4=898.53 eV)樣品大,表明Al2O3-CeO2-CP和Al2O3-CeO2-MR樣品中Al3+和Ce4+具有更強(qiáng)的相互作用。通過(guò)對(duì)比進(jìn)一步發(fā)現(xiàn),更大的結(jié)合能出現(xiàn)在Al2O3-CeO2-MR樣品中。在Al2O3-CeO2催化劑中浸漬和共沉淀使Ce的最外層電子向Al偏移,導(dǎo)致Ce的最外層電子云密度降低,電子結(jié)合能增大[33]。同時(shí)在微流控連續(xù)共沉淀過(guò)程中Al置換前驅(qū)體中的Ce,使Ce和Al的界面作用進(jìn)一步增強(qiáng)。
Al2O3-CeO2復(fù)合氧化物XPS的O 1s軌道圖譜如圖3(b)所示,結(jié)合能在 529.5~531.0 eV對(duì)應(yīng)于晶格氧(Olattic),結(jié)合能在531.0~532.5 eV歸屬于表面缺陷氧(Osur)[34]。結(jié)合表1中氧空位濃度Osur/(Olatt+Osur)數(shù)據(jù),Al2O3-CeO2-PM物混樣品氧空位濃度最低,為38.5%,浸漬和共沉淀樣品氧空位濃度均有所增加,分別為40.5%與42.7%,但最高的氧空位濃度出現(xiàn)在Al2O3-CeO2-MR復(fù)合氧化物中(46.4%),表明該樣品具有更多的氧空位缺陷。
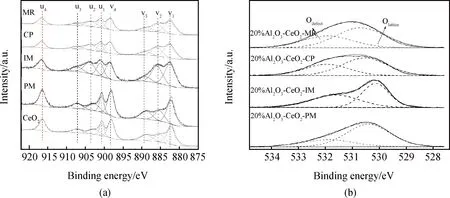
圖3 (a)不同Al2O3-CeO2復(fù)合氧化物Ce 3d軌道XPS圖譜;(b)不同Al2O3-CeO2復(fù)合氧化物的O 1s軌道XPS圖譜Fig.3 (a) Ce 3d XPS spectra of various Al2O3-CeO2 composite oxides; (b) O 1s XPS spectra of various Al2O3-CeO2 composite oxides
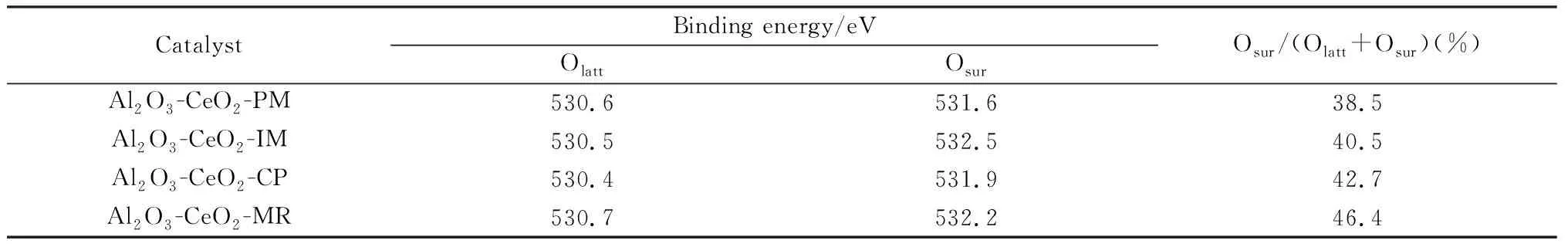
表1 不同Al2O3-CeO2復(fù)合氧化物的氧空位濃度Table 1 Oxygen vacancy concentration of various Al2O3-CeO2 composite oxides
由不同方法制備的Al2O3-CeO2復(fù)合氧化物N2吸附-脫附等溫曲線如圖4所示,比表面積和孔容見(jiàn)如表2所示。單一Al2O3、CeO2及復(fù)合氧化物均為介孔結(jié)構(gòu),明顯地,Al2O3-CeO2-PM物混樣品與單一的CeO2相似,具有Ⅳ型等溫線H2型回滯環(huán)。然而,Al2O3-CeO2-IM、Al2O3-CeO2-CP和Al2O3-CeO2-MR樣品卻具有典型的Ⅳ型等溫線H3型回滯環(huán),浸漬和共沉淀作用在一定程度上改變了介孔的孔形狀特征。由表2數(shù)據(jù)可知,單一介孔Al2O3與CeO2比表面積分別為201.2 m2/g、39.1 m2/g,Al2O3-CeO2復(fù)合氧化物的比表面積均介于兩者之間,在不同方法制得的復(fù)合氧化物中,Al2O3-CeO2-MR具有最大的比表面積140.2 m2/g和較小的孔容0.30 cm3/g。

表2 單一CeO2、Al2O3及不同Al2O3-CeO2復(fù)合氧化物的比表面積和孔結(jié)構(gòu)參數(shù)Table 2 Specific surface areas and pore structure parameters of sole CeO2, Al2O3 and various Al2O3-CeO2 composite oxides
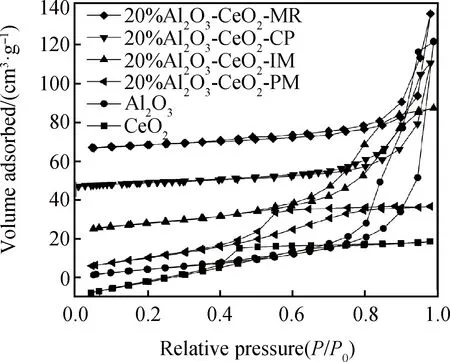
圖4 單一Al2O3、CeO2及Al2O3-CeO2復(fù)合氧化物N2等溫吸附-脫附曲線Fig.4 Adsorption-desorption isotherms of sole Al2O3, CeO2 and Al2O3-CeO2 composite oxides
由不同方法制備的Al2O3-CeO2復(fù)合氧化物SEM(見(jiàn)圖5)和TEM照片(見(jiàn)圖6)可知,不同樣品均為納米尺度晶粒堆積,Al2O3-CeO2-PM樣品表面呈大顆粒分布,分散性較差(見(jiàn)圖5(a))。Al2O3-CeO2-IM表面晶粒分布較少,呈現(xiàn)層狀分布(見(jiàn)圖5(b))。共沉淀法制備的Al2O3-CeO2-CP,表面晶粒分布并不均勻,存在團(tuán)聚堆積現(xiàn)象(見(jiàn)圖5(c)),晶粒尺寸集中在12~18 nm(見(jiàn)圖6(b))。而Al2O3-CeO2-MR樣品晶粒尺寸更小且分布更為集中(4~14 nm)(見(jiàn)圖6(d)),這與微流控工藝過(guò)程優(yōu)異的混合效率和傳質(zhì)傳熱特性有關(guān)。同時(shí),反應(yīng)過(guò)程中恒定的pH反應(yīng)體系,易于晶體的成核控制。而傳統(tǒng)共沉淀法極易出現(xiàn)濃度和溫度梯度,且過(guò)程中不穩(wěn)定的pH反應(yīng)體系促使反應(yīng)物顆粒易團(tuán)聚,不利于晶體成核。

圖5 不同Al2O3-CeO2復(fù)合氧化物的SEM照片F(xiàn)ig.5 SEM images of various Al2O3-CeO2 composite oxides
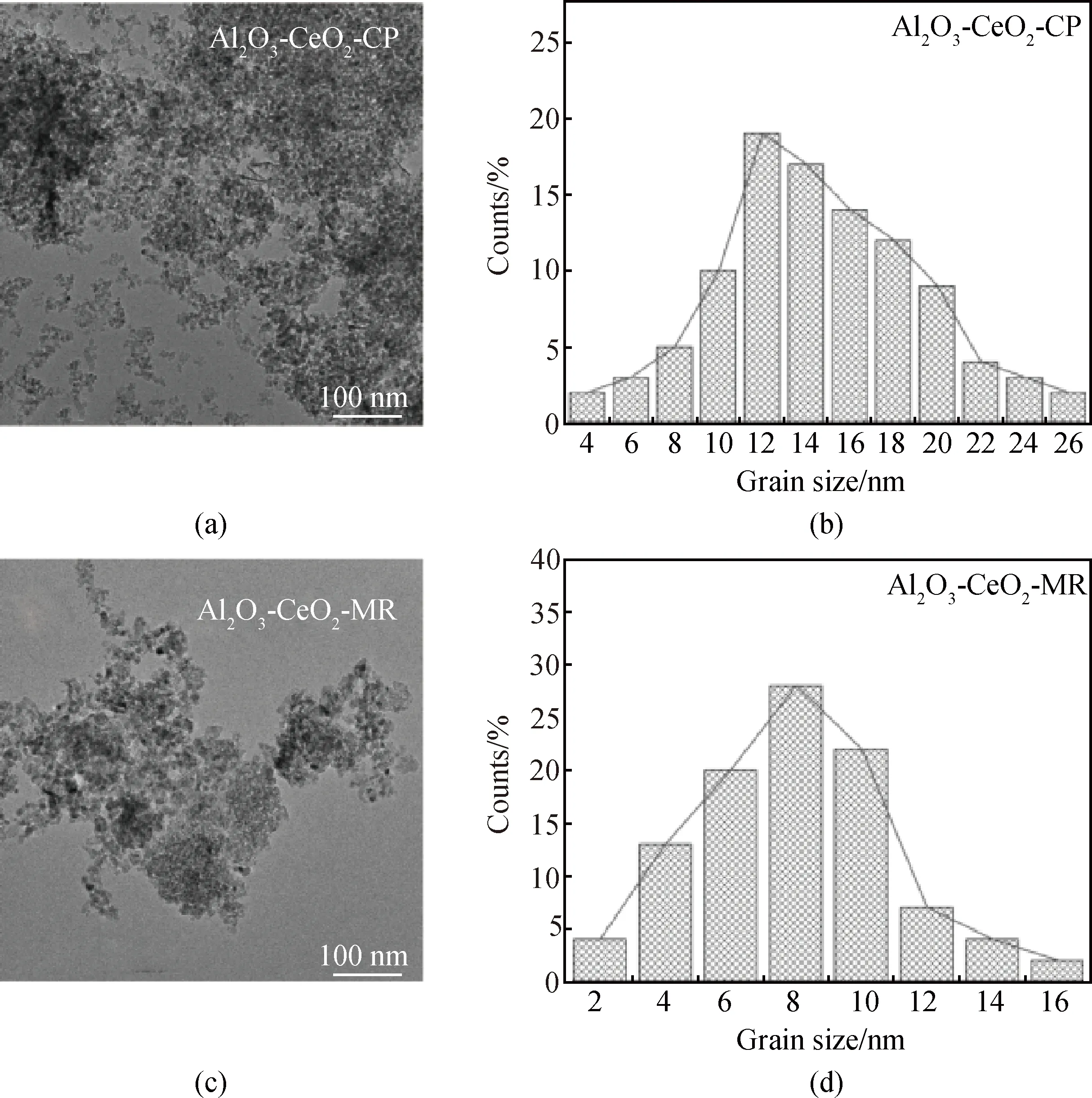
圖6 (a),(c)Al2O3-CeO2-CP和Al2O3-CeO2-MR復(fù)合氧化物的TEM照片;(b),(d)Al2O3-CeO2-CP和Al2O3-CeO2-MR的晶粒尺寸分布圖Fig.6 (a), (c) TEM images of Al2O3-CeO2-CP and Al2O3-CeO2-MR composite oxides; (b), (d) grain size distributions of Al2O3-CeO2-CP and Al2O3-CeO2-MR composite oxides
綜上分析,不同制備方法對(duì)Al2O3/CeO2復(fù)合相界面性質(zhì)影響較大。物混樣品Al2O3-CeO2-PM僅為簡(jiǎn)單機(jī)械混合,無(wú)特殊化學(xué)復(fù)合界面存在。以CeO2為載體,采用浸漬法制備的Al2O3-CeO2-IM形成了復(fù)合相界面,引起了該樣品部分電子結(jié)構(gòu)性質(zhì)的改變。程度更大的電子結(jié)構(gòu)性質(zhì)調(diào)變出現(xiàn)在共沉淀樣品中,共沉淀形成的特殊復(fù)合相界面能引起Al2O3-CeO2復(fù)合氧化物結(jié)合能的改變。然而,與傳統(tǒng)共沉淀樣品Al2O3-CeO2-CP相比,微流控連續(xù)共沉淀法制得的Al2O3-CeO2-MR晶粒尺寸更小且分布更為集中,Al2O3-CeO2-MR均勻的復(fù)合相界面產(chǎn)生了更多的氧空位缺陷。這也可從不同復(fù)合樣品對(duì)CO2吸附特性數(shù)據(jù)得到證實(shí),如圖7所示。不難看出,不同制備方法得到的樣品在80~150 ℃均表現(xiàn)出較強(qiáng)的CO2吸附能力(見(jiàn)圖7(a))。經(jīng)對(duì)比可以發(fā)現(xiàn),Al2O3-CeO2-PM與Al2O3-CeO2-IM樣品的CO2相對(duì)吸附量明顯較低,共沉淀制得的樣品Al2O3-CeO2-CP和Al2O3-CeO2-MR對(duì)CO2吸附表現(xiàn)出更好的吸附能力(見(jiàn)圖7(b)),其中,Al2O3-CeO2-MR具有最大的CO2吸附量,表現(xiàn)出潛在的CO2吸附活化能力。
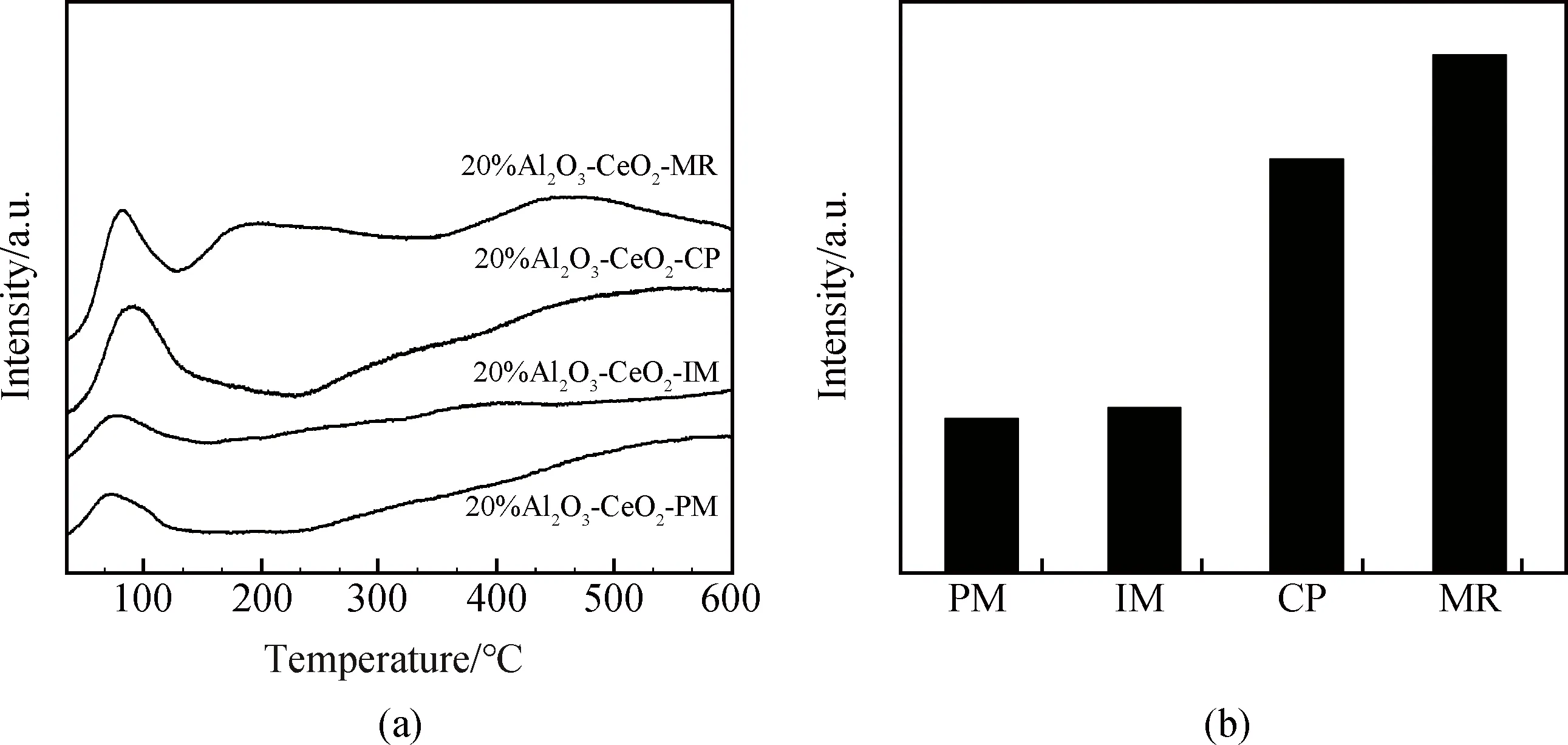
圖7 (a)不同Al2O3-CeO2復(fù)合氧化物的CO2-TPD圖;(b)不同Al2O3-CeO2復(fù)合氧化物的CO2相對(duì)吸附量Fig.7 (a) CO2-TPD profiles of various Al2O3-CeO2 composite oxides; (b) relative CO2 adsorptions of various Al2O3-CeO2 composite oxides
2.2 催化性能評(píng)價(jià)
在混合原料氣配比為V(H2)∶V(CO2)∶V(N2)=72∶24∶4、反應(yīng)溫度320 ℃、反應(yīng)壓力3 MPa、體積空速9 000 mL·g-1·h-1條件下,考察了不同方法制得Al2O3-CeO2復(fù)合氧化物對(duì)CO2轉(zhuǎn)化率、甲醇選擇性及甲醇時(shí)空產(chǎn)率的影響,并與單一Al2O3和CeO2催化性能進(jìn)行了比較,結(jié)果如圖8(a)所示。不難發(fā)現(xiàn),單一Al2O3和CeO2催化性能較差,二者的CO2轉(zhuǎn)化率分別僅為5.2%和6.5%,甲醇的選擇性僅為61.2%與72.2%。兩相復(fù)合后催化劑的催化性能得到不同程度的提升,Al2O3-CeO2-PM復(fù)合氧化物CO2轉(zhuǎn)化率、甲醇選擇性及甲醇時(shí)空產(chǎn)率分別為7.9%、78.5%和0.031 g·mL-1·h-1。然而,最佳的催化性能出現(xiàn)在Al2O3-CeO2-MR復(fù)合氧化物中,CO2轉(zhuǎn)化率、甲醇選擇性及甲醇時(shí)空產(chǎn)率分別達(dá)到15.3%、86.4%和0.076 g·mL-1·h-1。同時(shí),也將Al2O3-CeO2與另外兩類鈰基復(fù)合氧化物的催化性能進(jìn)行了比較,結(jié)果如圖8(b)所示,表明Al2O3-CeO2催化性能更優(yōu)。TiO2-CeO2、La2O3-CeO2的CO2轉(zhuǎn)化率分別為9.8%、12.3%,甲醇選擇性分別為79.5%、82.5%。同時(shí),Al2O3-CeO2-MR復(fù)合樣品在320 ℃的CO2轉(zhuǎn)化率(15.3%)與經(jīng)熱力學(xué)分析軟件(HSC-8)計(jì)算的CO2平衡轉(zhuǎn)化率理論值(14.7%)基本吻合(見(jiàn)圖8(a))。

圖8 (a)單一Al2O3、CeO2及不同Al2O3-CeO2復(fù)合氧化物的催化性能;(b)微流控連續(xù)共沉淀法制得TiO2-CeO2、La2O3-CeO2和Al2O3-CeO2的催化性能Fig.8 (a) Catalytic performance of sole Al2O3, CeO2 and various Al2O3-CeO2 composite oxides;(b) catalytic performance of TiO2-CeO2, La2O3-CeO2 and Al2O3-CeO2 prepared by microfluidic continuous coprecipitation
對(duì)Al2O3-CeO2-MR復(fù)合氧化物催化劑的反應(yīng)穩(wěn)定性進(jìn)行了測(cè)試(見(jiàn)圖9(a))。由圖可知,Al2O3-CeO2-MR復(fù)合催化劑在60 h范圍內(nèi)催化性能并無(wú)明顯衰減,表現(xiàn)出優(yōu)異的反應(yīng)穩(wěn)定性,甲醇選擇性穩(wěn)定在81%,CO2轉(zhuǎn)化率維持在11%以上。Al2O3-CeO2-MR復(fù)合氧化物的重復(fù)性實(shí)驗(yàn)結(jié)果如圖9(b)所示,催化劑的CO2轉(zhuǎn)化率和甲醇選擇性隨催化劑的使用次數(shù)增加,呈現(xiàn)出略微下降趨勢(shì)。Al2O3-CeO2-MR在第四次進(jìn)行評(píng)價(jià)之后,CO2轉(zhuǎn)化率仍達(dá)10.6%,甲醇選擇性為80.5%,Al2O3-CeO2-MR具有較好的重復(fù)性。
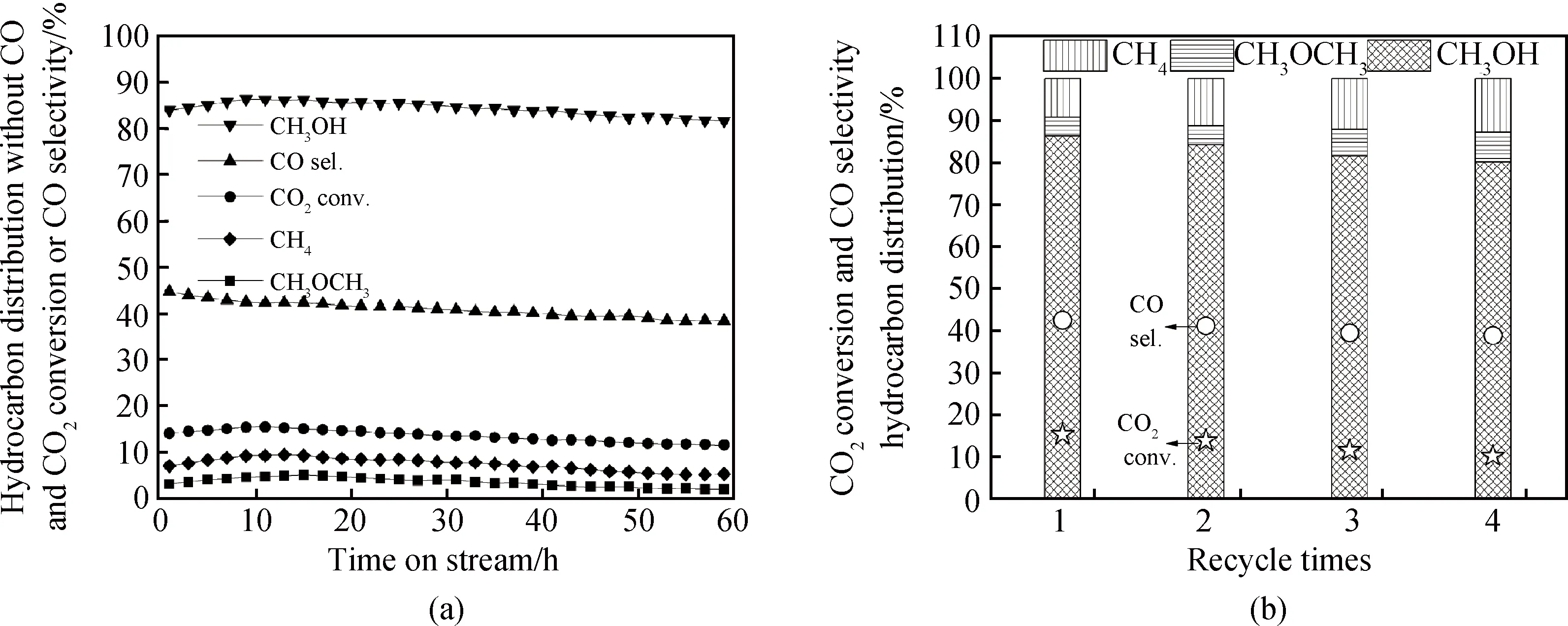
圖9 (a)Al2O3-CeO2-MR穩(wěn)定性測(cè)試;(b)Al2O3-CeO2-MR重復(fù)性實(shí)驗(yàn)Fig.9 (a) Stability test of Al2O3-CeO2-MR and (b) reproducibility performance of Al2O3-CeO2-MR
由反應(yīng)前后Al2O3-CeO2-MR的XPS圖譜(見(jiàn)圖10)可知,經(jīng)評(píng)價(jià)后催化劑Ce4+與Ce3+的特征峰強(qiáng)度減弱,對(duì)應(yīng)的結(jié)合能降低(見(jiàn)圖10(a)),同時(shí)氧空位缺陷濃度降至43.1%(見(jiàn)圖10(b)),表明反應(yīng)后Al2O3-CeO2-MR催化劑氧空位出現(xiàn)一定猝滅現(xiàn)象,導(dǎo)致其對(duì)CO2的吸附量降低(見(jiàn)圖11),進(jìn)一步表明氧空位缺陷濃度對(duì)CO2催化轉(zhuǎn)化具有促進(jìn)作用。結(jié)合復(fù)合氧化物的物化性質(zhì)分析結(jié)果,Al2O3-CeO2-MR樣品優(yōu)異的催化性能與形成的豐富氧空位缺陷密不可分。
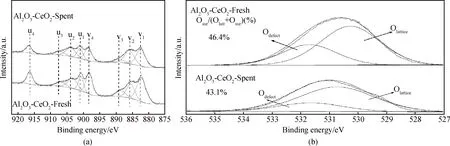
圖10 (a)反應(yīng)前后Al2O3-CeO2-MR的Ce 3d的XPS譜圖;(b)反應(yīng)前后Al2O3-CeO2-MR的氧空位缺陷濃度Fig.10 (a) Ce 3d XPS spectra of the fresh and spent Al2O3-CeO2-MR; (b) oxygen vacancy defect concentrations of the fresh and spent Al2O3-CeO2-MR
圖11 反應(yīng)前后Al2O3-CeO2-MR的CO2-TPD圖譜及CO2的相對(duì)吸附量Fig.11 CO2-TPD profiles and relative CO2 adsorption of the fresh and spent Al2O3-CeO2-MR
由上可知,Al2O3-CeO2-MR因形成特殊的復(fù)合界面相而產(chǎn)生的大量氧空位缺陷是該類樣品催化性能大幅提升的關(guān)鍵。然而,與傳統(tǒng)共沉淀相比,微流控連續(xù)共沉淀法具有不可比擬的優(yōu)勢(shì)。微流控技術(shù)制得樣品晶粒尺寸更小且分布更為集中,產(chǎn)生的氧空位缺陷更為豐富,在催化CO2加氫制甲醇中反應(yīng)性能更為優(yōu)異。
2.3 Al2O3-CeO2-MR復(fù)合氧化物的原位紅外分析
為研究Al2O3-CeO2-MR復(fù)合氧化物催化CO2加氫制甲醇的反應(yīng)機(jī)制,借助原位漫反射傅里葉變換紅外光譜對(duì)Al2O3-CeO2-MR催化反應(yīng)40 min的中間活性態(tài)進(jìn)行分析,結(jié)果如圖12所示。由圖可知,在2 901 cm-1處的紅外振動(dòng)峰歸屬于甲酸鹽HCOO*物種—CHO基團(tuán)上C—H鍵的彎曲振動(dòng),1 618 cm-1和1 378 cm-1處的紅外振動(dòng)峰分別反映甲酸鹽HCOO*物種—OCO基團(tuán)的非對(duì)稱與對(duì)稱伸縮振動(dòng)信息[24,35]。隨反應(yīng)時(shí)間從1 min增加到40 min,可以觀察到HCOO*中間體穩(wěn)定存在,表明Al2O3-CeO2-MR復(fù)合氧化物中氧空位缺陷有助于穩(wěn)定甲酸鹽中間體[36-37]。而2 837 cm-1處的紅外振動(dòng)對(duì)應(yīng)甲氧基CH3O*物種CH3O—基團(tuán)C—O鍵的伸縮振動(dòng)峰,2 983 cm-1紅外振動(dòng)峰對(duì)應(yīng)甲氧基CH3O*物種—CH3基團(tuán)上C—H鍵的伸縮振動(dòng)。可見(jiàn),在反應(yīng)過(guò)程中甲酸鹽物種(HCOO*)和甲氧基物種(CH3O*)同時(shí)存在,表明CO2加氫反應(yīng)在Al2O3-CeO2-MR復(fù)合氧化物上遵循甲酸鹽反應(yīng)機(jī)制。
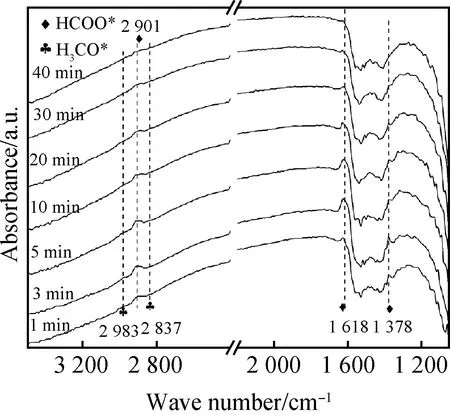
圖12 Al2O3-CeO2-MR復(fù)合氧化物的原位紅外圖譜(測(cè)試條件:V(H2)/V(CO2)/V(N2)=72/24/4,1 MPa,320 ℃,75 mL/min)Fig.12 In-situ infrared spectra of Al2O3-CeO2-MR composite oxide (Test conditions: V(H2)/V(CO2)/V(N2)=72/24/4, 1 MPa, 320 ℃, 75 mL/min)
圖13為Al2O3-CeO2-MR催化劑上CO2和H2吸附活化的反應(yīng)機(jī)理示意圖,H2吸附在CeO2晶體表面形成H2*,經(jīng)氫異裂解作用產(chǎn)生金屬Ce位點(diǎn)與O活性位點(diǎn)的HCe*和HO*,隨后金屬Ce位點(diǎn)HCe*轉(zhuǎn)移到鄰近的O活性位點(diǎn)形成HO*。CO2在復(fù)合氧化物的堿性位點(diǎn)被吸附后經(jīng)氧空位Al-□-Ce活化,再與活化的HO*結(jié)合成甲酸鹽中間體(HCOO*),HCOO*經(jīng)質(zhì)子化后形成H2COOH*,H2COOH*活性中間體裂解為甲醛基(H2CO*)和羥基(OH*),吸附的H2CO*基團(tuán)進(jìn)一步氫化成甲氧基(H3CO*),最終生成甲醇(CH3OH)目標(biāo)產(chǎn)物[38]。

圖13 Al2O3-CeO2-MR催化劑上CO2加氫制甲醇反應(yīng)機(jī)理Fig.13 Proposed reaction mechanism of CO2 hydrogenation to methanol over Al2O3-CeO2-MR
3 結(jié) 論
不同制備方法對(duì)Al2O3/CeO2復(fù)合界面性質(zhì)及催化CO2加氫制甲醇反應(yīng)具有重要影響。物混樣品只有兩相物理混合,無(wú)特殊化學(xué)復(fù)合界面的形成。浸漬樣品中Al2O3/CeO2界面出現(xiàn)一定的結(jié)構(gòu)性質(zhì)調(diào)變,但貧瘠的氧空位缺陷致使催化性能提高并不明顯。共沉淀樣品中Al2O3/CeO2界面結(jié)合能增大,界面相互作用增強(qiáng),形成的復(fù)合界面產(chǎn)生了大量氧空位缺陷,在CO2活化轉(zhuǎn)化過(guò)程中表現(xiàn)出優(yōu)異的催化性能和較好的穩(wěn)定性。更為重要的是,微流控連續(xù)共沉淀法在復(fù)合氧化物制備方面具有不可比擬的優(yōu)勢(shì)。獨(dú)特的微通道結(jié)構(gòu)使其具有優(yōu)異的混合效率和高效的傳質(zhì)傳熱效率,為兩相復(fù)合提供了恒定的反應(yīng)條件和快速的反應(yīng)路徑,制得的Al2O3-CeO2復(fù)合氧化物具有更為豐富的氧空位缺陷,這是CO2加氫制甲醇獲得較高CO2轉(zhuǎn)化率和甲醇選擇性的關(guān)鍵。原位紅外分析表明微流控連續(xù)共沉淀法制備的Al2O3-CeO2-MR復(fù)合氧化物遵循甲酸鹽反應(yīng)機(jī)制,而氧空位缺陷有助于穩(wěn)定甲酸鹽反應(yīng)中間體。
猜你喜歡
當(dāng)代陜西(2020年13期)2020-08-24 08:22:02
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
制造技術(shù)與機(jī)床(2017年5期)2018-01-19 02:49:17
金秋(2017年4期)2017-06-07 08:22:16
中國(guó)材料進(jìn)展(2016年10期)2016-12-26 06:50:20
濰坊學(xué)院學(xué)報(bào)(2016年2期)2016-12-01 13:00:11
浙江大學(xué)學(xué)報(bào)(工學(xué)版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國(guó)資源綜合利用(2016年4期)2016-01-22 08:27:23
