用CRISPR-Cas9在綿羊成纖維細胞ACTG1導入熒光標記基因
2021-11-04 09:05:12郭延華皮文輝
新疆農業科學 2021年9期
關鍵詞:檢測
郭延華,皮文輝
(綿羊遺傳改良與健康養殖國家重點實驗室/新疆農墾科學院畜牧獸醫研究所,新疆石河子 832000)
0 引 言
1 材料與方法
1.1 材 料
pX330-U6-Chimeric_BB-CBh-hSpCas9(Addgene,42230),pCAS9-mCherry-Frame+1(Addgene,66940),pCRISPaint-TagGFP2-PuroR(Addgene,80970),pMAX(VDC-1040,Lonza)電轉儀(Amax NucleofectorⅡ),流式細胞儀(BDFACSAria Ⅲ)。綿羊原代成纖維細胞采自新疆農墾科學院畜牧獸醫研究所種羊場,采集8月齡試驗羊只耳緣組織,帶回實驗室原代培養保存。
QuickExtract DNA 抽提液(QE09050,Epicentre)、PrimerSTAR DNA Polymerase(R045,Takara)、Surveyor 凝膠電泳突變檢測試劑盒(706025,Transgenomic)、10× T4ligase buffer(B0202S,NEB)、T4PNK(M0201S,NEB)、2X rapid ligase buffer(B1010,Enzymatics)、T7ligase(L6020L,Enzymatics)和FastDigestBbsI(FD1014,Fermentas)。
細胞培養皿、離心管(Nunc);DMEM 高糖培養液(61965059,Gibco);胎牛血清(LOT1407738,BI);胰酶替代物 TrypLETMExpress(1X)(12604-021,Gibco)。
1.2 方 法
1.2.1 gRNAs(guide RNAs)設計
針對綿羊ACTG1基因TAA終止密碼子位點,3’端保留PAM(NGG)。表1

表1 綿羊ACTG1基因羧基端gRNAs序列Table 1 Oligo sequences of C-terminal targets of sheep ACTG1 gene
AGCAGGAGTATGATGAGTCCACCGCAAATGCTTCTAAAGTGGATCAGCAAGCAGGAGTATGATGAGTCTGGCCCCTCCATTGTCCACCGCAAATGCTTCTAAACGGACTGCGAGCAGACAAATGCTTCTAAACGGACC
1.2.2 gRNA質粒克隆
王積薪手捻一粒黑玉棋子停了下來,一張白玉般的臉變成了醬紫色,半晌抬頭對星雨道:“那夜月光入戶,照在床枕邊,徘徊轉移,有一刻就停留在她的臉上。”
磷酸化退火gRNA寡核苷酸,BbsI酶切pX330-U6-Chimeric_BB-CBh-hSpCas9質粒,用Golden Gate克隆體系,將退火寡核苷酸連接,構建gRNA質粒。首先將合成的正向和反向gRNA寡核苷酸用水稀釋至100 μm,分別取1與6.5 μL的水混合,加入10×T4ligase buffer 1 μL和T4PNK 0.5 μL,將10 μL液體混勻,水浴37℃ 30 min。然后將反應管轉入PCR儀,95℃ 3 min,直接關閉PCR儀,保持關閉PCR儀頂蓋,室溫下自動降溫退火30 min。將雙鏈退火的反應液補加90 μL水稀釋10倍,混勻代用。Golden Gate反應體系是:2×rapid ligase buffer 5 μL、BSA(20 mg/mL)0.3 μL、FastDigest BbsI 0.5 μL、T7 ligase 0.5 μL、退火稀釋的gRNA溶液1 μL、pX330-U6-Chimeric_BB-CBh-hSpCas9(25 ng/μL)1 μL和水2.0 μL,混勻。37℃ 水浴60 min。取2~5 μL 連接反應液,直接轉染50 μL感受態Stbl3。抗性Amp的LB固體培養基涂板,37℃ 16 h培養。
1.2.3 細胞轉染
采用電轉染綿羊成纖維細胞。待細胞生長至培養皿的80%,消化收集2×105個細胞,20 μL電轉液[7]懸浮,加入相應的質粒,將電擊杯放置于電轉儀的電擊槽中,運行CZ-167電轉程序,培養液懸浮細胞后鋪于6孔板中培養。
1.2.4 CRISPR-Cas9系統活性檢測
檢測3個gRNA質粒編輯效率,電轉染20 μL,添加gRNA質粒1 μg。培養72 h收集細胞,用30 μL QuickExtract DNA 抽提液裂解細胞,水浴65℃ 10 min;95℃ 15 min。獲得的細胞裂解液作為基因組DNA模板,ShACTG1-F:AGCATGACTGACCTCCCTTTG 和shACTG1-R:CCCAACCCCATGTAAGACCG引物,primeSTAR酶擴增,98℃ 10s,60℃ 10s,72℃ 5s(38個循環);72℃ 2 min;25℃ ∞。電泳切膠回收后,按照Surveyor 凝膠電泳突變檢測試劑盒說明進行檢測。
1.2.5 流式細胞檢測
ACTG1基因標記組的細胞,電轉染20 μL體系添加的質粒是:250 ng基因組編輯的gRNA質粒、250 ng供體模板gRNA質粒pCAS9-mCherry-Frame+1、500 ng供體模板質粒pCRISPaint-TagGFP2-PuroR。同時單獨電轉500 ng供體模板質粒pCRISPaint-TagGFP2-PuroR樣品,觀察細胞熒光。電轉陽性細胞樣品250 ng紅色熒光蛋白(red fluorescent protein,RFP)質粒pCAS9-mCherry-Frame+1和250 ng綠色熒光蛋白(green fluorescent protein,GFP)質粒pMAX,作為流式細胞儀紅色熒光蛋白和綠色熒光蛋白細胞標準。電轉染細胞培養72 h,用熒光顯微鏡觀察成纖維細胞GFP和RFP表達。然后收集細胞。重懸在0.5 mL的PBS中,流式細胞儀分析(BDFACSAriaⅢ,FACSDiva Version 6.1.3)。FITC通道檢測GFP細胞,PE通道檢測RFP細胞。每份樣品細胞計數超過10 000個。
1.2.6 單克隆篩選
電轉染20 μL體系添加的質粒是250 ng pX330-Cas9-ACTG1-2質粒、250 ng pCAS9-mCherry-Frame+1質粒、500 ng模板質粒pCRISPaint-TagGFP2-PuroR,培養60 h,加入嘌呤霉素1 μg/mL培養液,持續培養5 d,直至培養皿底部形成細胞克隆。用PBS漂洗2遍。然后加入8倍稀釋的PBS胰酶替代物消化液,室溫靜置7 min。在體式顯微鏡下用口吸管吸取單克隆細胞。轉入96孔板中繼續培養,經過1次傳代培養,直至長滿3孔,收集用于檢測。
1.2.7 測 序
將收集的嘌呤霉素抗性篩選的單克隆細胞,用30 μL QuickExtract DNA 抽提液裂解細胞,水浴65℃ 10 min;95℃ 15 min。獲得的細胞裂解液作為基因組DNA模板,ShACTG1-F和shACTG1-R引物,primeSTAR酶擴增,98℃ 10s,60℃ 10s,72℃ 6 min(38個循環);72℃ 10 min;25℃ ∞。電泳切膠回收后,PCR產物直接送樣測序,測序引物是ShACTG1-F 和shACTG1-R。
2 結果與分析
2.1 綿羊成纖維細胞ACTG1基因終止密碼子附近CRISPR-Cas9的gRNA靶向活性
綿羊成纖維細胞ACTG1基因終止密碼子附近設計的3個gRNA位點,電轉染導入CRISPR-Cas9表達系統質粒,培養72 h后。圖1
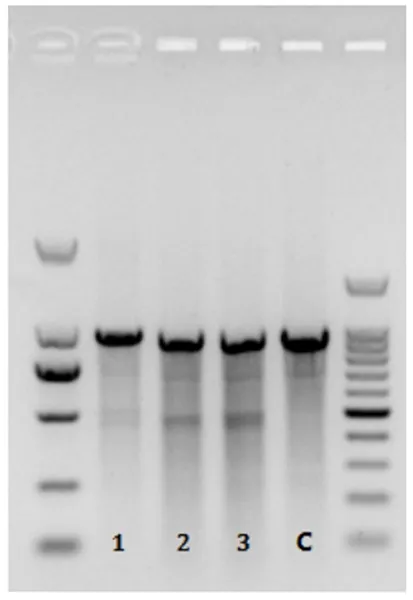
圖1 Surveyor檢測CRISPR-Cas9系統的電泳圖Fig.1 CRISPR-Cas9 surveyor detection gel electrophoresis map
1、2和3號泳道樣本同正常細胞C泳道樣品比較,存在不同的DNA異源雙鏈雜交酶切帶型,說明3個設計的gRNA靶點,采用CRISPR-Cas9系統,在綿羊成纖維細胞的基因組ACTG1基因終止密碼子靶點附近,成功誘導產生DNA雙鏈斷裂,經細胞修復后產生突變。
2.2 流式細胞儀檢測
熒光顯微鏡觀察標記實驗組,看到有RFP細胞和紅綠兩色的熒光細胞,初步判定GFP基因標記成功。熒光顯微鏡觀察供體模板質粒pCRISPaint-TagGFP2-PuroR的細胞樣品,未見到GFP。流式細胞儀檢測坐標圖顯示,Q1區域是RFP細胞,Q2區域是RFP+GFP雙色熒光細胞,Q4區是GFP細胞。pX330-Cas9-ACTG1-1、pX330-Cas9-ACTG1-2和pX330-Cas9-ACTG1-3質粒電轉標記組,紅綠雙色熒光細胞比率占整個檢測細胞分別是0.4%、1.4%、0.8%。圖2
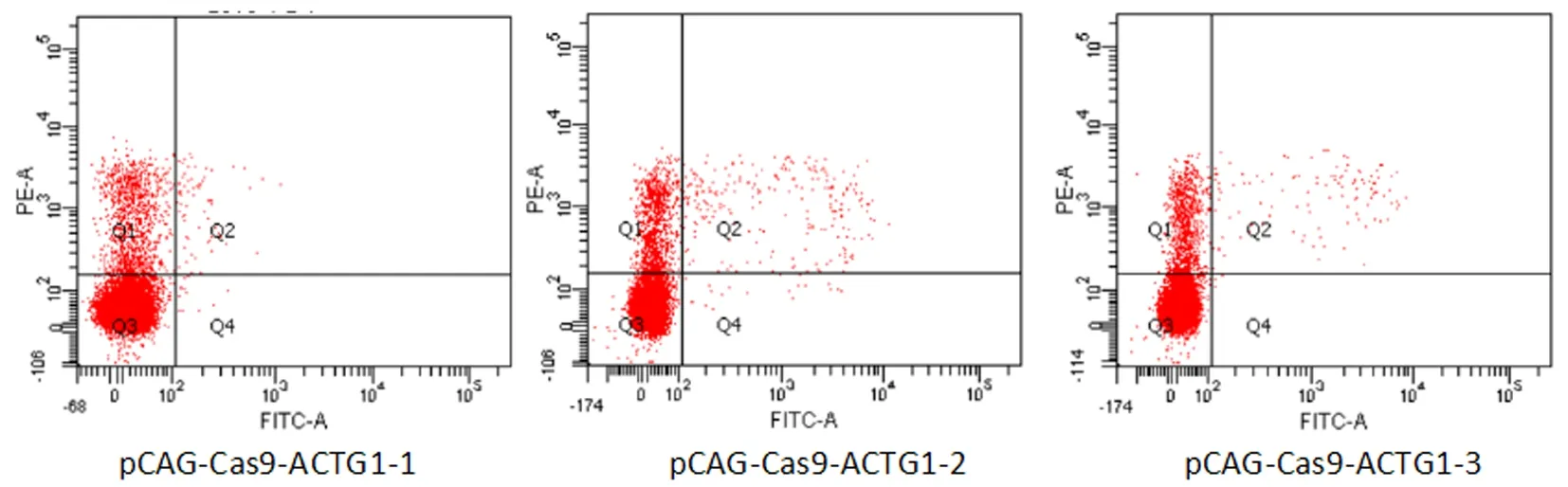
注:Q1(左上象限)為Y軸陽性細胞;Q2(右上象限)為雙陽性細胞;Q3(左下象限)為雙陰性細胞;Q4(右下象限)為X軸陽性細胞Note:圖2 流式細胞儀檢測綿羊ACTG1羧基端標記效率Fig.2 FACS analysis and efficiency of C-terminal tag GFP in sheep ACTG1 gene
由于pCAS9-mCherry-Frame+1質粒表達RFP,流式細胞儀檢測計數的RFP細胞數量(Q2+Q1),就是pCAS9-mCherry-Frame+1質粒在綿羊成纖維細胞中的轉染效率。實驗采用的轉染體系,流式細胞儀檢測結果顯示,pCAS9-mCherry-Frame+1質粒在綿羊成纖維細胞轉染效率達4%~7.5%。而pMAX細胞樣品組,流式細胞儀檢測的轉染效率達40%。轉染效率差異顯著的主要原因是2個質粒大小差異顯著,pMAX(3 486 bp)和pCAS9-mCherry-Frame+1(10 122 bp)。
2.3 PCR擴增檢測
終止密碼子5’連接端,ShACTG1-F測序結果比較分析顯示,整合的外源基因片段,消除了終止密碼子。缺失和插入堿基數都是3連密碼子整數倍,整體序列沒有發生移碼,編碼框可以正常編碼GFP密碼子。篩選得到克隆細胞株都實現了GFP基因編碼框的正確閱讀,可以正常轉錄翻譯表達蛋白,這也符合抗性標記嘌呤霉素選擇的結果。圖3
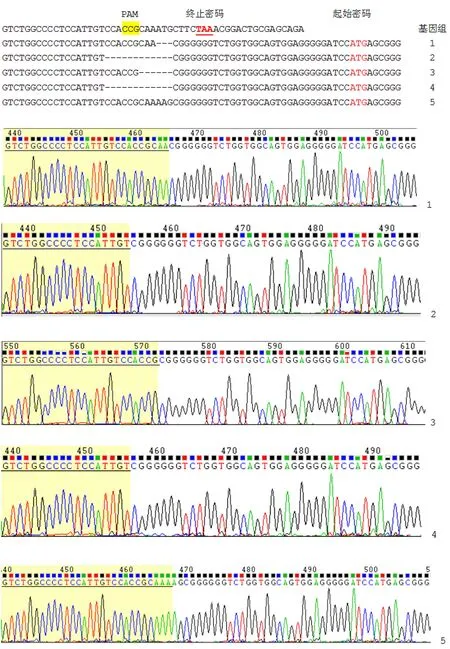
圖3 ACTG1羧基端標記5’測序結果比對Fig.3 Comparison of 5 'sequencing results of ACTG1 C-terminal tag GFP
終止密碼子3’連接端,ShACTG1-R測序結果比較分析顯示,抗性標記選擇的細胞克隆,整合了不同長度的供體模板骨架。紅色字母是綿羊細胞基因組序列,下劃線黑斜體字母是pCRISPaint-TagGFP2-PuroR骨架序列。后面數字是3’端堿基對應的供體模板質粒骨架上的位置。圖4
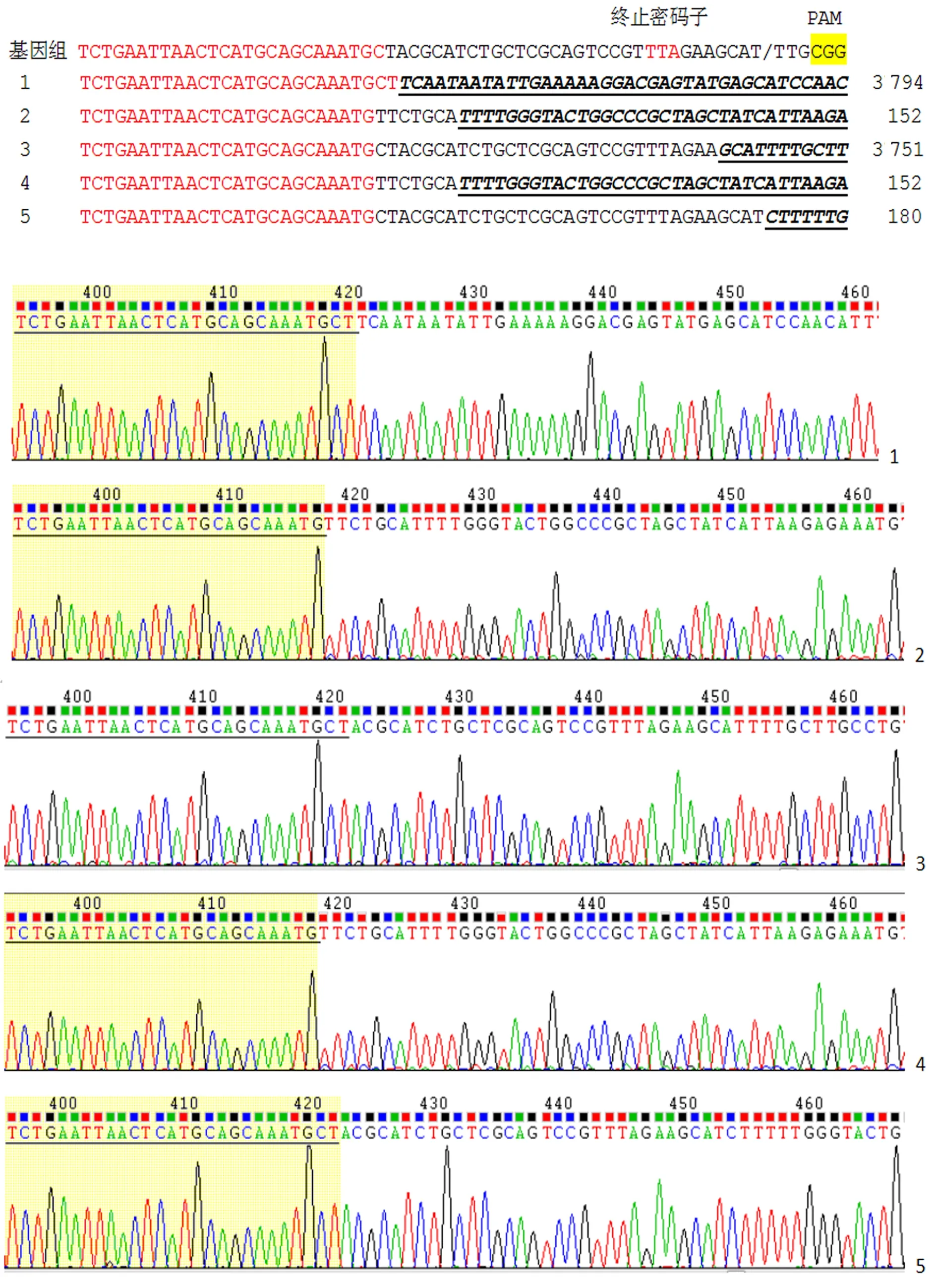
圖4 ACTG1羧基端標3’測序結果比對Fig.4 Comparison of 3 'sequencing results of ACTG1 C-terminal tag
3 討 論
人工核酸內切酶(Engineered endonuclease,EEN)定點切割基因組,產生特定位點基因組雙鏈斷裂(Double-strand brakes,DSBs)或切口(Nicks),為剖析生物體的基因功能研究和應用,開辟了新的方式[1,2,8,9,10]。
提高外源基因精確導入基因組的效率,是基因組編輯重要應用之一[11]。基因導入起源于基因打靶技術[12]。基因打靶技術利用了HR修復途徑,將外源DNA片段精確導入靶位點。基因打靶技術效率極其低,主要是因為HR修復途徑在細胞內采用頻率低。當EEN在細胞內誘導產生DSB后,存在3條可能的修復途徑:HR、NHEJ和MMEJ[13]。NHEJ修復途徑在細胞內采用頻率非常高,為了獲得高的基因導入效率,NHEJ修復途徑是一個較好的選擇。通過NHEJ修復途徑,獲得了較高的基因導入效率[14-19]。在細胞內,利用EEN同時在活細胞的基因組和供體模板中誘導DSB,通過NHEJ修復途徑,使得線性化供體模板和切割的基因組之間發生連接。在斑馬魚和植物中,敲入效率達30%[17,19]。
CRISPaint技術是模塊化基因標記系統,利用CRISPR-Cas9系統,在細胞內共同實現基因組和供體模板不同靶點斷裂,通過NHEJ修復途徑,將供體模板導入基因組特定位點[3,5]。利用CRISPaint技術,在綿羊成纖維細胞ACTG1基因羧基端標記GFP效率達1.4%。通過嘌呤霉素選擇,獲得了基因標記成功的單克隆細胞。
測序嘌呤霉素選擇獲得的單克隆細胞,顯示NHEJ連接修復呈現不同序列,缺失和插入堿基數都是3連密碼子整數倍,整體序列沒有發生移碼,編碼框可以正常編碼GFP和嘌呤霉素抗性蛋白。NHEJ修復連接產生很多結果,而那些發生移碼的修復細胞,在嘌呤霉素選擇下將無法生存。單克隆細胞3’端連接,呈現不同的骨架片段,說明線性化供體模板在細胞內受多種因素作用,發生不同位點斷裂。
利用CRISPR-Cas9系統,結合MMEJ修復途徑,也實現了高效的基因組定點整合外源基因[20,21,22]。該方法由于需要微同源臂,實施起來需要針對不同位點構建包含微同源臂質粒。CRISPaint方法利用NHEJ修復途徑,包含大量模塊化質粒。如果是標記或干擾目的基因,無需構建質粒。CRISPR-Cas9系統,結合NHEJ精確導入外源基因,是一種高效便利的技術[3,4]。
4 結 論
CRISPaint通過cNHEJ在CRISPR-Cas9作用的細胞中整合異源遺傳物質,提供了一種簡單、精確和快速的方法[3]。利用CRISPaint能夠快速有效的將大的外源DNA序列插入綿羊成纖維細胞的預定基因組位點。針對綿羊ACTG1基因終止密碼子位點,導入熒光標記基因,流式細胞儀檢測,整合效率達1.4%。同時,pCAS9-mCherry-Frame+1質粒在綿羊成纖維細胞轉染效率是4%~7.5%,說明10 kb左右大小的Cas9相關質粒,轉染效率較低。因此,提高CRISPaint系統的Cas9相關質粒轉染效率,將改善外源基因定點整合進入綿羊成纖維細胞基因組的效率。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
