超高效液相色譜-串聯(lián)質(zhì)譜法測(cè)定菊花15種吡咯里西啶生物堿毒素
2021-11-05 13:59:32章豪吳銀良朱勇趙健陳國
浙江農(nóng)業(yè)科學(xué) 2021年11期
章豪,吳銀良,朱勇,趙健,陳國
(寧波市農(nóng)業(yè)科學(xué)研究院 農(nóng)業(yè)農(nóng)村部農(nóng)產(chǎn)品質(zhì)量安全風(fēng)險(xiǎn)評(píng)估實(shí)驗(yàn)室(寧波),浙江 寧波 315040)
吡咯里西啶生物堿(PAs)是植物抵御昆蟲及食草動(dòng)物等侵害的一類化學(xué)物質(zhì),廣泛分布在植物中[1]。目前,已在菊科的植物中檢測(cè)到PAs[2]。聯(lián)合國糧農(nóng)組織和世界衛(wèi)生組織(WHO)認(rèn)為,PAs是草藥產(chǎn)品與食品中最廣泛和最嚴(yán)重的摻雜性與內(nèi)源性的毒性成分[3]。PAs的毒性與結(jié)構(gòu)密切相關(guān),可形成烯丙酯結(jié)構(gòu)的PAs具有肝臟毒性,會(huì)造成肝硬化、肝細(xì)胞壞死等癥狀[4]。歐盟于2019年實(shí)施PAs限量規(guī)定,成人每天攝入量不超過0.35 μg,兒童不超過0.14 μg[5]。WTO貿(mào)易技術(shù)壁壘委員會(huì)曾因我國出口的某種藥材中PAs含量過高而發(fā)布通告禁令;歐盟食品和飼料類快速預(yù)警系統(tǒng)也曾因我國出口至歐盟的草本茶原料中PAs含量過高發(fā)布警告。我國法定藥材標(biāo)準(zhǔn)中已收載了菊花等多種含有PAs的藥材,但對(duì)PAs在菊花中的含量及種類等缺乏相關(guān)的數(shù)據(jù)。目前,我國尚未建立高效的檢測(cè)方法,因此,還未能準(zhǔn)確評(píng)估其在菊花中的含量水平,亟須對(duì)我國常見菊花中PAs的檢測(cè)方法進(jìn)行研究,進(jìn)一步通過摸底檢測(cè)明確菊花中PAs含量數(shù)據(jù),以期厘清我國菊花食品安全風(fēng)險(xiǎn),確定關(guān)鍵控制點(diǎn),形成管控措施,應(yīng)對(duì)潛在的國際貿(mào)易壁壘風(fēng)險(xiǎn)。
目前,針對(duì)PAs殘留采用的分析方法主要為液相色譜-串聯(lián)質(zhì)譜法(LC-MS/MS)[6-9]、液相色譜法(LC)[10-12]、氣相色譜-質(zhì)譜法(GC-MS)[13-15]、氣相色譜法(GC)[16-19]、分光光度法[20]、核磁共振法[21]和免疫學(xué)方法[22]。但使用分光光度法、核磁共振法和免疫學(xué)方法時(shí)存在基體干擾大的問題,更多是用來篩選,難以對(duì)多種PAs毒素進(jìn)行分析。而GC和GC-MS分析PAs存在過程煩瑣、耗時(shí)長(zhǎng)、穩(wěn)定性差、檢出限高等缺陷,PAs分析使用 GC及GC-MS 較少,LC或LC-MS/MS是PAs主要分析手段。超高效液相色譜-串聯(lián)質(zhì)譜法(UPLC-MS/MS)在 PAs 分析中選擇性好、靈敏度高,且適用于同時(shí)檢測(cè)多種 PAs,因而廣泛用于農(nóng)產(chǎn)品或食品中PAs 的定性定量分析[9]。
PAs是含有特征性堿性氮的還原性生物堿,通常使用半極性或極性有機(jī)溶劑或在酸化水條件下萃取,其氧化形式(PANOs)屬于極性分子,容易被極性溶劑(例如甲醇)或稀酸水溶液萃取[6]。PAs前處理技術(shù)主要包括液液萃取(LLE)法、固相萃取(SPE)法、QuEChERS 法,以及發(fā)展的新型技術(shù)如分散液相微萃取(DLLME)、固相支持液/液萃取(SLE)[6-19]。其中,SPE是常用凈化PAs的前處理方法。常用固相萃取柱有C18和C8柱[8-11]。它們存在共提物多的缺點(diǎn),在色譜分析中存在大的背景干擾,會(huì)對(duì)定量分析的準(zhǔn)確性以及靈敏度產(chǎn)生影響[7]。對(duì)于生物堿的凈化,陽離子交換柱是一種更為有效的純化方法,可以有效地去除弱極性與離子型極性干擾物質(zhì),對(duì)PAs及其氮氧化物均有良好的純化效果,并且不受其他共流出物的干擾[8]。
我們通過優(yōu)化儀器條件和樣品前處理方法,建立了基于Oasis MCX固相萃取柱能同時(shí)對(duì)菊花15種PAs毒素殘留進(jìn)行UPLC-MS/MS分析的方法。該方法具有精密度和準(zhǔn)確度高、檢測(cè)耗時(shí)短的特點(diǎn),能同時(shí)對(duì)菊花樣品多種PAs毒素進(jìn)行測(cè)定。
1 材料與方法
1.1 材料
UPLC XevoTMTQ-S micro 超高效液相色譜-串聯(lián)質(zhì)譜儀,配有電噴霧離子源(ESI)及MassLynxV4.1數(shù)據(jù)處理系統(tǒng)(美國Waters公司);低溫高速離心機(jī)(德國Sigma公司);Vortex Genie 3 漩渦振蕩器(德國IKA公司)。
PAs標(biāo)準(zhǔn)品(天芥菜堿、天芥菜堿-N-氧化物、倒千里光堿、倒千里光堿-N-氧化物、千里光寧堿、千里光寧堿-N-氧化物、千里光堿、千里光堿-N-氧化物、促黑激素、促黑激素-N-氧化物、千里光菲靈堿、千里光菲靈堿-N-氧化物、歐天芥菜堿、歐天芥菜堿-N-氧化物和克氏千里光堿)純度大于95%(德國PhytoLab GmbH & Co. KG公司);甲酸(色譜純,美國Tedia公司);乙腈、甲醇(色譜純,德國Merck公司);Oasis MCX固相萃取小柱(200 mg/6 mL,美國Waters公司);實(shí)驗(yàn)用水為Milli-Q超純水。
1.2 方法
1.2.1 標(biāo)準(zhǔn)溶液配制
分別準(zhǔn)確稱取適量(精確至0.1 mg)15種PAs標(biāo)準(zhǔn)品,用甲醇溶解,配制質(zhì)量濃度為1 g·L-1的標(biāo)準(zhǔn)儲(chǔ)備液,于-20 ℃ 保存;分別準(zhǔn)確吸取1 mL標(biāo)準(zhǔn)儲(chǔ)備液,置于100 mL容量瓶中,用甲醇稀釋,配制質(zhì)量濃度為10 mg·L-1的混合標(biāo)準(zhǔn)儲(chǔ)備液,于-20 ℃保存。
基質(zhì)匹配標(biāo)準(zhǔn)溶液。制備菊花空白樣品溶液,樣品處理方法采用1.2.2節(jié),通過稀釋混合標(biāo)準(zhǔn)儲(chǔ)備液配制基質(zhì)匹配標(biāo)準(zhǔn)溶液。
1.2.2 樣品處理
稱取5.00 g(精確至0.01 g)菊花樣品置于50 mL離心管中,加入10 mL 0.1 mol·L-1硫酸水溶液,10 000 r·min-1均質(zhì)1 min,超聲提取15 min;以5 000 r·min-1速度離心10 min,將上清液收集,殘?jiān)儆?0 mL 0.1 mol·L-1硫酸水溶液提取1次,合并提取液。分別采用5 mL甲醇與5 mL去離子水活化Oasis MCX固相萃取小柱,移取提取液至固相萃取小柱,待液面到達(dá)柱床表面時(shí)用5 mL水淋洗,再用1%甲酸水溶液淋洗,最后用5 mL 0.5%氨水甲醇溶液洗脫,洗脫液在40 ℃下氮?dú)獯蹈桑挥? mL 0.1%(V/V)甲酸水-甲醇(90∶10,V/V)溶液復(fù)溶,過0.22 μm濾膜,供UPLC-MS/MS分析。
1.2.3 色譜條件
色譜柱:Acquity UPLC BEH C18(1.7 μm,100 mm×2.1 mm);流動(dòng)相:A相為0.1%(V/V)甲酸水溶液,B相為甲醇;梯度洗脫程序:0~1.0 min,10%B;1.0~7.0 min,10%B~90%B;7.0~8.0 min,90%B;8.0~8.1 min,90%B~10%B;8.1~10.0 min,10%B;流速0.30 mL·min-1;進(jìn)樣量為10 μL。
1.2.4 質(zhì)譜條件
多反應(yīng)離子監(jiān)測(cè)(MRM)模式;ESI源正離子模式電離;錐孔氣流速:50 L·h-1;脫溶劑氣流速:1 000 L·h-1;脫溶劑氣溫度:500 ℃;離子源溫度:150 ℃;毛細(xì)管電壓:2.0 kV;萃取錐孔電壓:20 V;RF透鏡電壓:0.5 V;15種PAs毒素的母離子、子離子及其錐孔電壓和碰撞能量見表1。
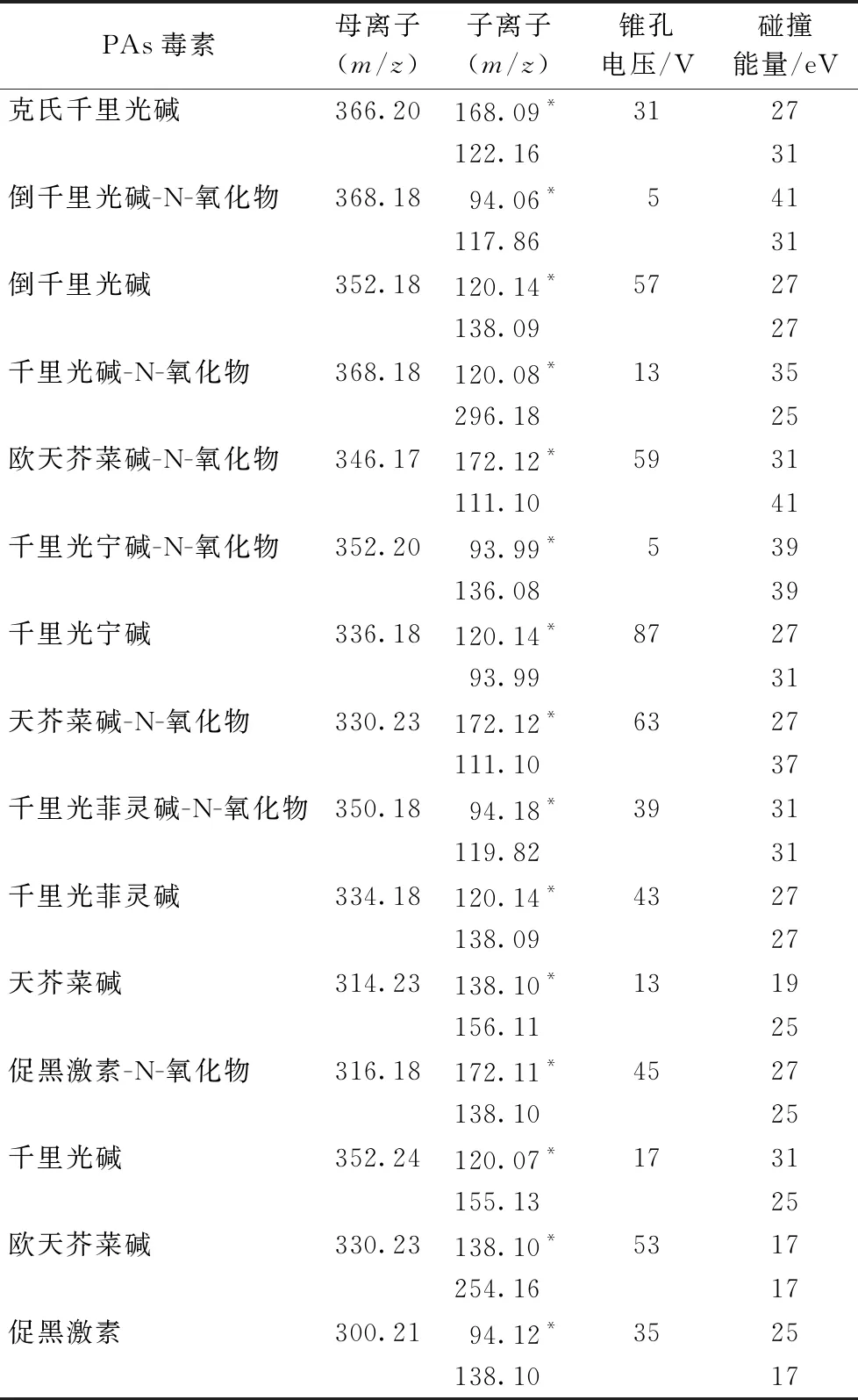
表1 15種PAs毒素的母離子、子離子及其錐孔電壓和碰撞能量
2 結(jié)果與分析
2.1 儀器條件優(yōu)化
在使用液相色譜-串聯(lián)質(zhì)譜法中多采用ESI源正離子電離模式來測(cè)定PAs毒素殘留[6-9]。采用甲醇分別配制1.0 mg·L-115種PAs毒素標(biāo)準(zhǔn)溶液,通過Intellistart軟件分別摸索15種PAs毒素的母離子、子離子及其錐孔電壓和碰撞能量(表1),15種PAs毒素在ESI源正離子電離模式下均可獲得較好的響應(yīng)。
考查乙腈和甲醇作為色譜流動(dòng)相的有機(jī)相時(shí)PAs毒素的分離效果,發(fā)現(xiàn)當(dāng)采用乙腈作為有機(jī)相時(shí),千里光堿、千里光菲靈堿-N-氧化物和倒千里光堿-N-氧化物的響應(yīng)較差,而采用甲醇作為有機(jī)相時(shí),15種PAs毒素的響應(yīng)均較好,因此,確定甲醇為有機(jī)相。
2.2 前處理?xiàng)l件優(yōu)化
PAs的提取,溶劑一般選用極性較大的溶液如甲醇[6-7]或水[8-9]等,加入少量的酸,可提高PAs的水溶性[14-17]。因此,本研究使用甲醇、水和0.05 mol·L-1硫酸水溶液3種溶劑對(duì)菊花樣品在10 μg·kg-1加標(biāo)水平下進(jìn)行提取。發(fā)現(xiàn)不加酸提取時(shí),采用水提取所有PAs毒素的回收率均低于采用甲醇提取的回收率;而加酸提取時(shí),采用0.05 mol·L-1硫酸水提取所有PAs毒素的回收率均高于采用甲醇提取的回收率,因此,確定以0.05 mol·L-1硫酸水溶液進(jìn)行提取。這可能是因?yàn)镻As毒素以離子形式存在于酸性環(huán)境中,顯著增強(qiáng)其水溶性,導(dǎo)致酸水的提取效率比甲醇高。
2.3 基質(zhì)效應(yīng)、標(biāo)準(zhǔn)曲線、檢出限和定量限
在考查菊花基質(zhì)中15種PAs毒素的基質(zhì)效應(yīng)時(shí)我們采用基質(zhì)匹配標(biāo)準(zhǔn)曲線的斜率/溶劑標(biāo)準(zhǔn)曲線的斜率×100%的計(jì)算方法。結(jié)果越接近100%,表明基質(zhì)效應(yīng)越小。當(dāng)結(jié)果小于100%時(shí)表明存在基質(zhì)抑制效應(yīng),大于100%時(shí)表明存在基質(zhì)增強(qiáng)效應(yīng)。表2顯示,15種PAs毒素均存在一定的基質(zhì)抑制效應(yīng)或是增強(qiáng)效應(yīng)。為了消除基質(zhì)效應(yīng)所帶來的影響,我們采用基質(zhì)匹配標(biāo)準(zhǔn)曲線來對(duì)目標(biāo)化合物進(jìn)行測(cè)定。采用菊花空白基質(zhì)液配制1~1 000 μg·L-1橫跨3個(gè)數(shù)量級(jí)的系列基質(zhì)標(biāo)準(zhǔn)工作溶液(其中千里光堿、千里光菲靈堿-N-氧化物和倒千里光堿-N-氧化物為5~1 000 μg·L-1)進(jìn)行UPLC-MS/MS測(cè)定分析,通過峰面積對(duì)濃度作標(biāo)準(zhǔn)曲線,從而得到線性回歸方程。15種PAs毒素在相應(yīng)濃度范圍內(nèi)相關(guān)系數(shù)均大于0.999,線性關(guān)系良好。通過3倍信噪比(S/N=3)進(jìn)行估算,15種PAs毒素的檢出限為0.5~9.0 μg·kg-1;通過10倍信噪比(S/N=10)進(jìn)行估算,15種PAs毒素的定量下限為1.7~30.0 μg·kg-1。
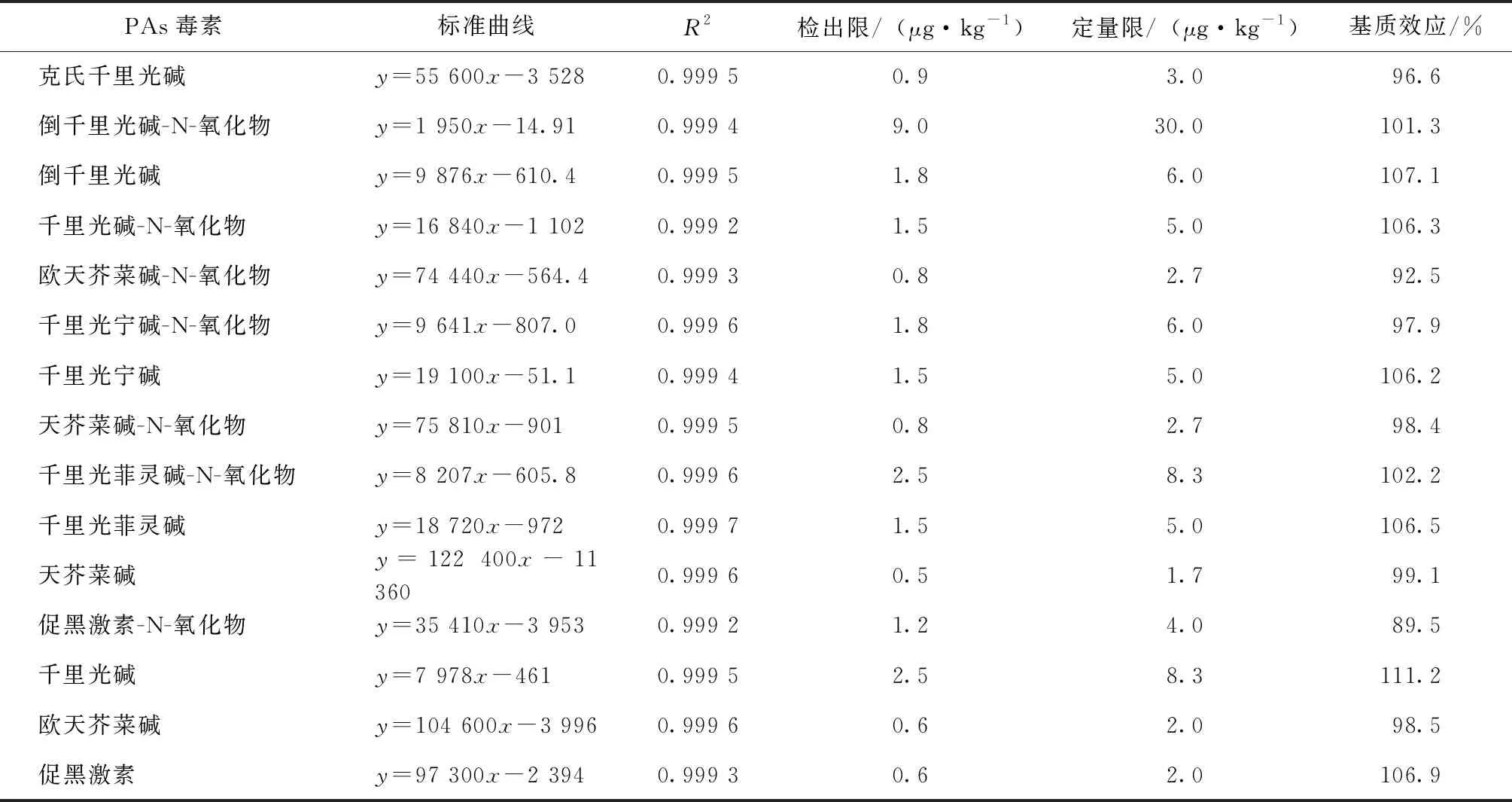
表2 15種PAs毒素在杭白菊基質(zhì)中的標(biāo)準(zhǔn)曲線、相關(guān)系數(shù)、檢出限、定量限和基質(zhì)效應(yīng)
2.4 回收率與精密度
對(duì)杭白菊進(jìn)行添加回收試驗(yàn),并計(jì)算相應(yīng)的相對(duì)標(biāo)準(zhǔn)偏差。杭白菊空白樣品中PAs毒素的加標(biāo)MRM色譜圖如圖1所示。杭白菊中千里光堿、千里光菲靈堿-N-氧化物和倒千里光堿-N-氧化物的添加量為30 μg·kg-1,其余12種PAs毒素的添加量為6 μg·kg-1,每批次同一濃度5個(gè)平行樣品,重復(fù)測(cè)定3次。

圖1 杭白菊加標(biāo)樣品的MRM色譜
表3顯示,15種PAs毒素的加標(biāo)回收率為82.6%~94.2%,相對(duì)標(biāo)準(zhǔn)偏差為2.3%~5.0%。
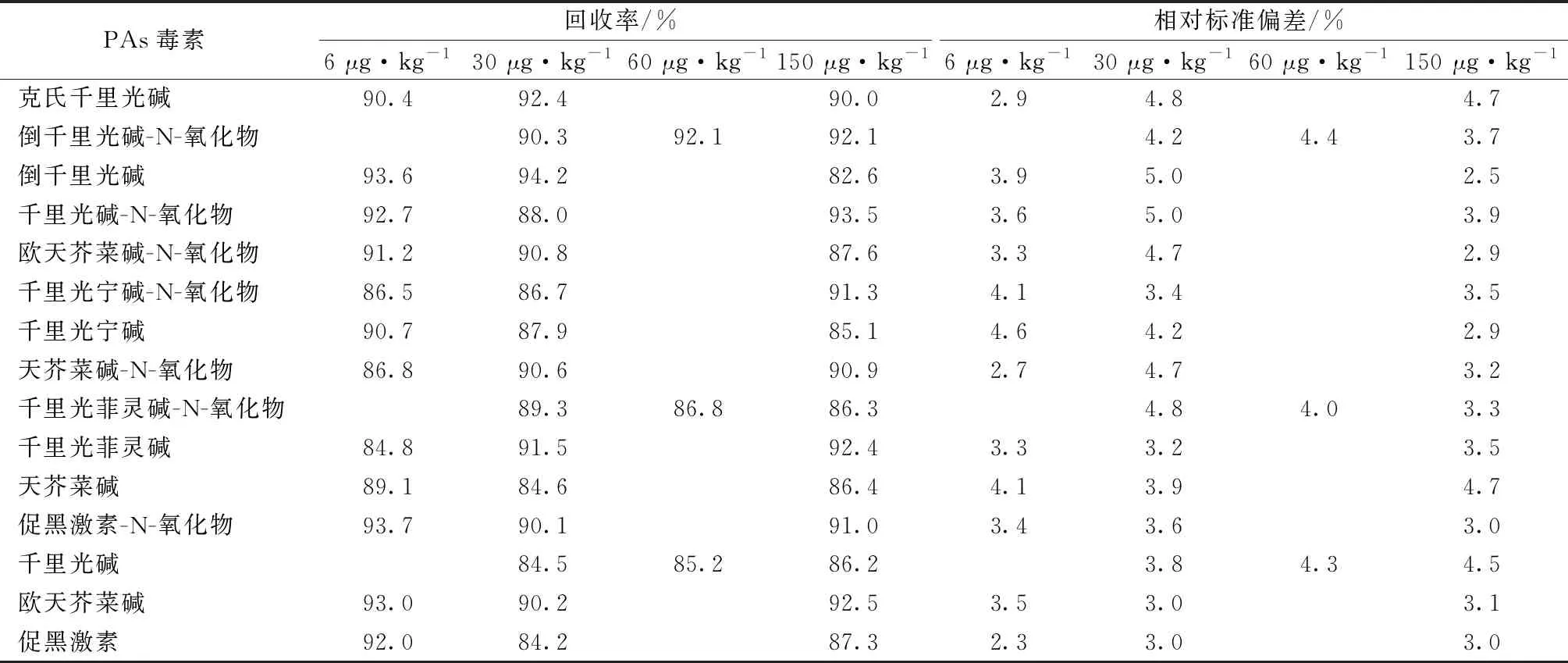
表3 15種PAs毒素在杭白菊中的回收率和相對(duì)標(biāo)準(zhǔn)偏差(n=5)
結(jié)果表明,方法對(duì)于15種PAs毒素在杭白菊中的殘留分析均具有較好的精密度和準(zhǔn)確度。
3 小結(jié)
建立了同時(shí)對(duì)菊花15種PAs毒素殘留進(jìn)行測(cè)定的UPLC-MS/MS方法,采用硫酸溶液提取菊花樣品中PAs毒素,離心后將上清液過Oasis MCX固相萃取小柱凈化,氮?dú)獯蹈珊髲?fù)溶,采用UPLC-MS/MS進(jìn)行分析。15種PAs毒素的添加回收率為82.6%~94.2%,相對(duì)標(biāo)準(zhǔn)偏差為2.3%~5.0%;檢出限為0.5~9.0 μg·kg-1,定量下限為1.7~30.0 μg·kg-1。該方法檢測(cè)耗時(shí)短,具有較高的精密度和準(zhǔn)確度,具備同時(shí)測(cè)定菊花樣品中多種PAs毒素的能力。
