氣相色譜-三重四極桿質譜法測定生濕面制品中單辛酸甘油酯的研究
2021-12-15 07:43:40季葛振
糧食與食品工業 2021年6期
陳 彬,季葛振,楊 俊,*,朱 云
1南通市食品藥品監督檢驗中心 (南通 226006)2南通市經濟技術開發區市場監督管理局 (南通 226009)
生濕面制品是指經過壓延成型并未經干燥處理的面制品,由于含水量高,若不進行保鮮處理則極易生長微生物,并腐敗變質[1],引起腐敗的主要微生物是細菌和霉菌[2-4]。單辛酸甘油酯作為一種新型無毒高效廣譜防腐劑,對細菌和霉菌均有抑制效果[5]。它在人體內和脂肪一樣,能分解代謝,無任何積蓄和不良反應。中國與世界各國和組織對于單甘油酯類的使用都有相關的規定,GB 2760—2011規定:“單辛酸甘油酯常用作面制品防腐劑用于濕切面,其最大使用量為1.0 g/kg”。
目前,單辛酸甘油酯的檢驗方法主要有氣相色譜法[6]和單四級桿氣質聯用法[7],而三重四級桿氣質聯用法檢測單辛酸甘油酯的方法未見報道。本文建立了氣相色譜-三重四極桿質譜測定食品中單辛酸甘油酯的方法,處理方法簡單高效,適用于生濕面制品中單辛酸甘油酯的測定。
1 材料與方法
1.1 儀器與試劑
7890B型氣相色譜儀,7000D型三重四極桿質譜儀,美國Agilent公司;乙腈,色譜純,德國Merk公司;氯化鈉,分析純,國藥集團;無水硫酸鈉(使用前在650 ℃灼燒 4 h ,貯于干燥器中,冷卻后備用),分析純,國藥集團;單辛酸甘油酯標準品,99.0%,美國NU-CHEK-PREP公司;Milli-Q高純水,由Millipore純水儀制備。
單辛酸甘油酯標準儲備液:稱取單辛酸甘油酯標準品1 mg,用丙酮溶解并定容至10 mL,配制成100 μg/mL的標準儲備液。
單辛酸甘油酯標準工作液:分別吸取單辛酸甘油酯標準儲備液0.01 mL、0.05 mL、0.10 mL、0.50 mL、1.00 mL于10 mL容量瓶中,用空白基質溶液定容至刻度,此工作液濃度為分別為0.1 μg/mL、0.5 μg/mL、1.0 μg/mL、5.0 μg/mL、10.0 μg/mL。
1.2 儀器條件
1.2.1氣相色譜條件
色譜柱1為HP-5石英毛細管柱(15 m×0.25 mm,0.25 μm),色譜柱2為HP-5石英毛細管柱(15 m×0.25 mm,0.25 μm),色譜柱1和色譜柱2串接,兩柱間連接反吹氣路;進樣口溫度300 ℃;脈沖分流進樣,脈沖壓力50 psi,脈沖時間為0.2 min,分流比為(5∶1),進樣量1 μL;載氣、反吹氣為高純氦氣;色譜柱1流量1.0 mL/min,色譜柱2流量1.2 mL/min;升溫程序為初始溫度150 ℃保持10 min,以20 ℃/min升至280 ℃,保持5 min。
1.2.2質譜條件
離子源:電子轟擊源,70 eV;離子源溫度280 ℃,接口溫度280 ℃;淬滅氣(高純氦氣)流量為2.25 mL/min,碰撞氣(高純氮氣)流量為1.5 mL/min。掃描方式為MRM多反應監測,單辛酸甘油酯離子對及碰撞能量見表1。
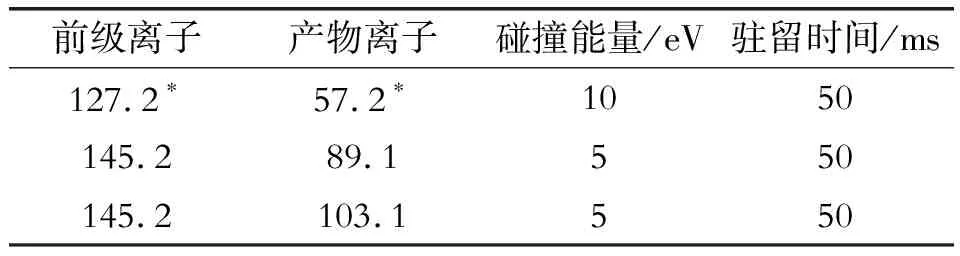
表1 單辛酸甘油酯離子對和碰撞能量
1.3 樣品前處理
稱取2 g樣品,加入10 mL乙腈+水(1+1,v/v)溶液10 mL,10 000 r/min勻漿1 min,加入2 g氯化鈉,渦旋混合,6 000 r/min離心5 min,取上層清液,加入1 g無水硫酸鈉,渦旋1 min,經0.22 μm有機濾膜過濾,按儀器工作條件進行測定。
2 結果與討論
2.1 色譜條件的優化
根據文獻資料,單辛酸甘油酯沸點較高,活性較大,在襯管中容易產生吸附。試驗選擇UI超高惰性襯管,并設置進樣口溫度為300 ℃,色譜柱起始溫度為150 ℃,降低單辛酸甘油酯在進樣環節產生的吸附。
試驗考察了脈沖分流(5∶1)和脈沖不分流兩種進樣方式對單辛酸甘油酯峰形的影響,結果表明:脈沖不分流進樣方式,響應值高,但峰形較差,容易產生拖尾。脈沖分流(5∶1)進樣加快單辛酸甘油酯進入色譜柱的速度,同時減少該物質進入色譜柱的量,所得峰形對稱,響應值適中,見圖1。試驗選擇脈沖分流(5:1)進樣,脈沖壓力50 psi,脈沖時間0.2 min。
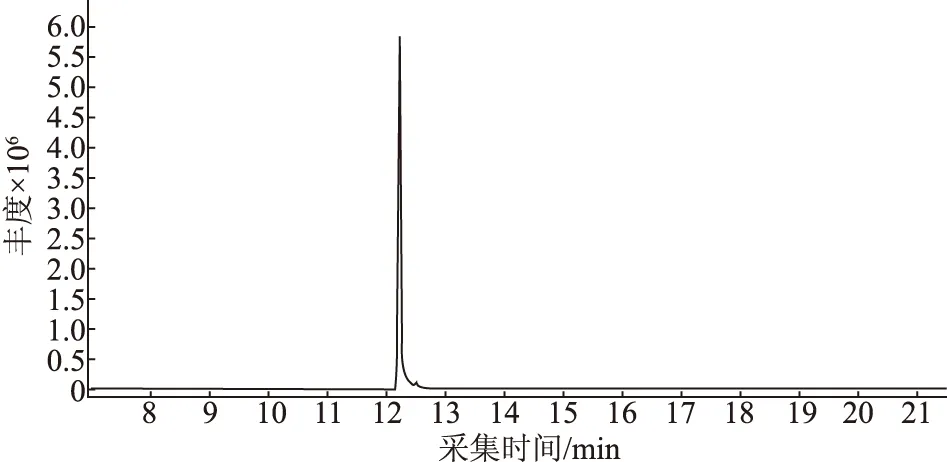
圖1 單辛酸甘油酯總離子流圖
2.2 質譜條件的優化
采用全掃描采集方式掃描50~300 amu范圍內單辛酸甘油酯質譜圖,見圖2。根據質譜圖中離子的豐度和質量數,為提高方法的靈敏度和選擇性,確定特征離子m/z 127.2、m/z 145.2作為前級離子。

圖2 單辛酸甘油酯全掃描質譜圖
選擇高純氮氣作為碰撞氣、高純氦氣作為淬滅氣,并分別設置氣流量為1.5 mL/min和2.25 mL/min,采用產物離子掃描方式,選擇并豐度較大的離子對m/z 127.2~57.2作為定量離子,m/z 145.2~89.1和m/z 145.2~103.1作為定性離子,見圖3、圖4。按照歐盟的法規,試驗選擇了兩個母離子和三個子離子,滿足4點鑒定法要求。
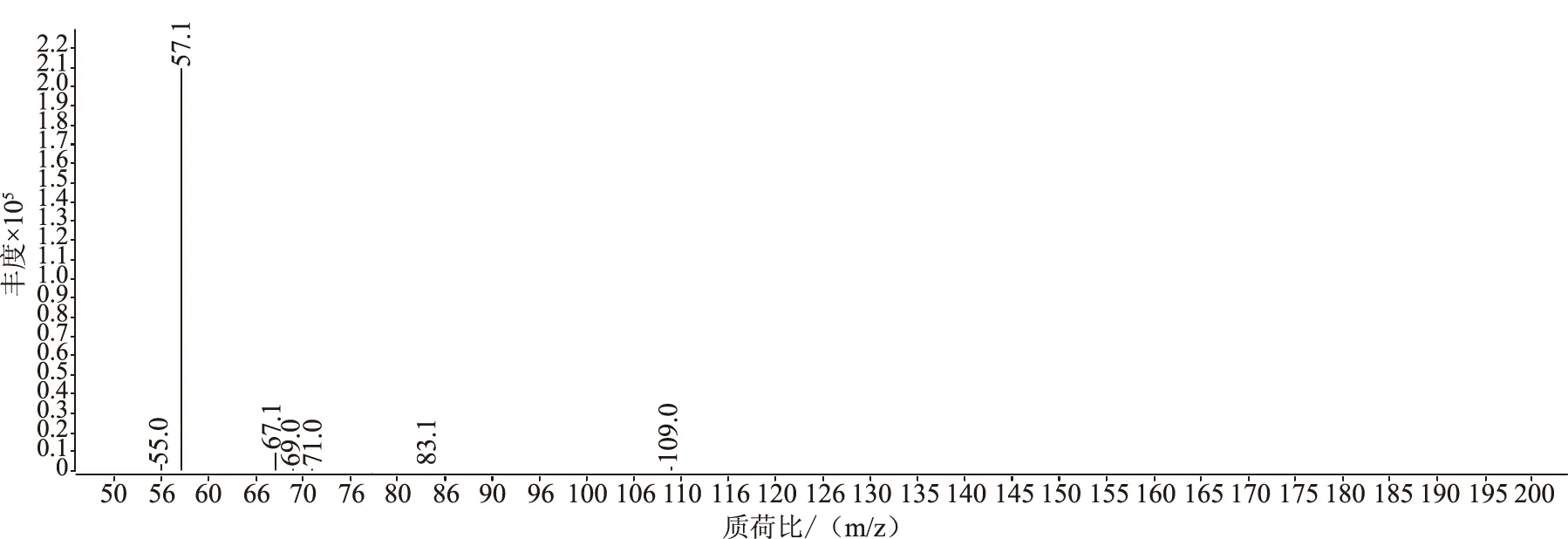
圖3 前級離子m/z 127.2二級全掃描質譜圖
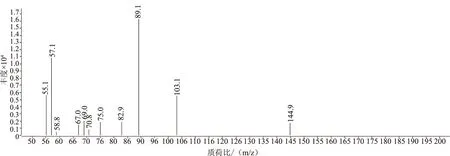
圖4 前級離子m/z 145.2二級全掃描質譜圖
根據選擇的離子對,試驗考察了碰撞能量為5、10、15、20 eV下,m/z 127.2~57.2、m/z 145.2~89.1和m/z 145.2~103.1三組離子對的響應值。結果表明:m/z 127.2~57.2在10 eV下有較好的響應,m/z 145.2~89.1和m/z 145.2~103.1在5 eV下有較好的響應。因此,在上述碰撞能量下,以m/z 127.2~57.2作為基峰,m/z 145.2~89.1和m/z 145.2~103.1相對于基峰的比率分別為13.2%和4.2%,輔助定性,見圖5。
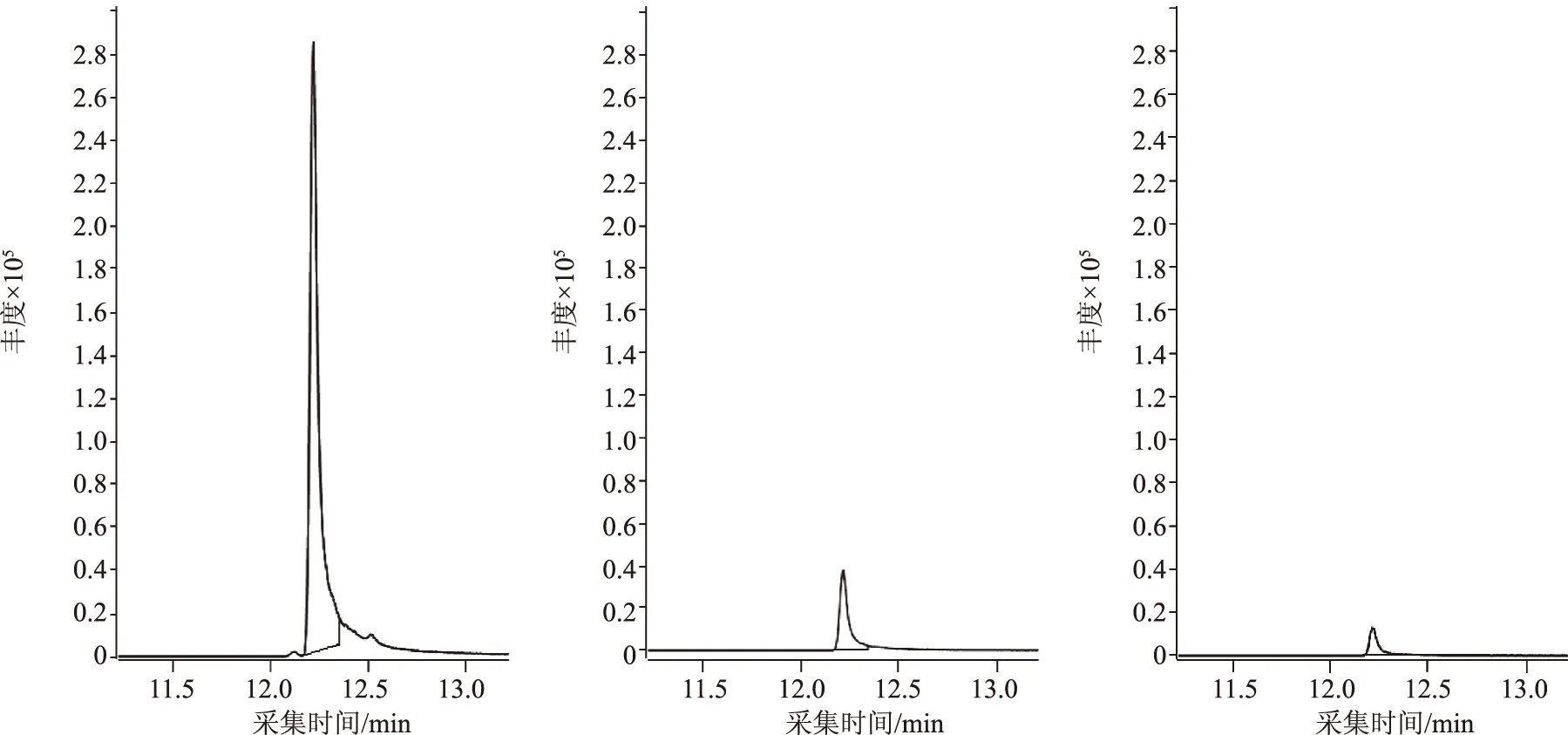
圖5 單辛酸甘油酯定量和定性離子對
2.3 樣品前處理條件的優化
單辛酸甘油酯極性較強[6],根據相似相溶原理,選擇強極性溶劑乙腈、水作為提取溶劑。試驗比較了乙腈、乙腈+水((1+1,v/v)的提取效果,結果表明:乙腈+水(1+1,v/v)滲透性能好,能進入生濕面制品內部,有利于充分提取樣品中的單辛酸甘油酯。
2.4 線性范圍和檢出限
試驗考察了樣品的基質效應,結果表明:生濕面制品有較強的基質增強效應,且能較好地改善峰形,提高靈敏度。使用空白基質溶液配制標準曲線:0.1、0.5、1.0、5.0、10.0 μg/mL。以m/z 127.2~57.2離子對峰面積為縱坐標,基質標準溶液濃度為橫坐標繪制標準工作曲線,得出線性方程和相關系數,單辛酸甘油酯在該濃度范圍內有良好的線性關系,其相關系數不小于0.998。在空白生濕面制品中加入將一定質量濃度的標準品,使樣品中的加標量為25 μg/kg。經過前處理,經儀器分析后,單辛酸甘油酯m/z 127.2~57.2定量離子信噪比大于3,因此,本試驗檢出限為25 μg/kg。
2.5 回收率和精密度
選擇餃子皮空白樣品,以10、25、50 mg/kg 3個水平進行添加回收試驗。每個水平做6次平行試驗,計算方法的回收率和精密度。平均回收率分別為108.2%、92.1%、80.5%,相對標準偏差(RSD)分別為5.2%、3.3%、7.8%。結果表明:該方法回收率穩定,數據精密度高,能滿足日常檢驗中單辛酸甘油酯準確定量的要求。
3 結論
本文建立了生濕面制品中單辛酸甘油酯的檢測方法,選用乙腈+水(1+1,v/v)混合溶劑高速勻漿提取樣品中單辛酸甘油酯,優化了氣相色譜、三重四級桿質譜儀的各項參數,采用脈沖分流(5:1)進樣方式,選擇m/z 127.2~57.2作為定量離子,m/z 145.2~89.1和m/z 145.2~103.1作為定性離子,兩對定性離子對相對于m/z 127.2~57.2的比率分別為13.2%和4.2%。本方法處理方法簡單高效,定性定量準確,靈敏度高,回收率高,重現性好,適用于生濕面制品中單辛酸甘油酯的測定。
