超高效液相色譜法同時測定障眼明膠囊中7個成分的含量
2021-12-21 10:17:10王月清
食品與藥品 2021年6期
王月清
(東南大學附屬中大醫院江北院區藥劑科,江蘇 南京 210048)
障眼明膠囊由石菖蒲、決明子、車前子、菟絲子、葛根、白芍、山茱萸、菊花、關黃柏、青葙子、甘草、黃芪、枸杞子、黨參、肉蓯蓉、升麻、蕤仁(去內果皮)、川芎、蔓荊子、黃精、熟地黃、密蒙花22位藥材組成,具有退翳明目,補益肝腎的功效,臨床主要用于初期及中期老年性白內障[1]。該藥收載于《新藥轉正標準》第82冊,質量標準含量測定項下采用高效液相色譜法(HPLC)測定芍藥苷含量,鑒別項下僅對關黃柏、甘草及黃芪做了定性檢查,目前文獻僅有障眼明膠囊中葛根素和芍藥苷的成分分析[2],未見其他藥材成分分析的相關報道。藥食同源的決明子自古有清熱明目作用,主要成分蒽醌類占1 %,典型化合物為橙黃決明素和大黃酚[3-4];車前子主要治療目赤腫痛、目視昏暗,其藥理活性成分主要為苯乙醇苷類毛蕊花糖苷及環烯醚萜類京尼平苷酸,《中國藥典》2020年版也將這兩種成分作為含量測定的指標[5-7];被《神農本草經》列為上品的菟絲子具有明目養肝,抑制白內障形成的功效,金絲桃苷是其主要活性成分之一[8-9];葛根能改善眼功能,白芍養陰平肝[10-11]。由于該中成藥由20味中藥組成,多指標的定量測定方法雖然成本較高,但更有利于對藥品質量的全面客觀的評價。本研究采用梯度洗脫法結合分段變波長檢測法,首次建立了同時測定橙黃決明素、大黃酚、毛蕊花糖苷、京尼平苷酸、金絲桃苷、葛根素、芍藥苷的超高效液相色譜(UPLC)法,結果表明,該方法簡便快速、靈敏度高,為該藥的質量控制提供了依據。
1 儀器與材料
1.1 儀器
安捷倫1290 infinity超高效液相色譜儀(包含二元梯度泵、自動進樣器、柱溫箱和PDA檢測器,安捷倫科技公司);ABS-135S型電子天平(d=0.01 mg,瑞士梅特勒公司);TGL-16G離心機(上海安亭科學儀器廠)。
1.2 材料
障眼明膠囊(批號:20190922,20191027,20191125,20191219,規格:0.25 g/粒,湖南德康制藥股份有限公司);橙黃決明素對照品(批號:111900-201605,純度:98.3 %),大黃酚對照品(批號:110796-201922,純度:99.4 %),毛蕊花糖苷對照品(批號:111530-201914,純度:95.2 %),京尼平苷酸對照品(批號:111828-201805,純度:98.1 %),金絲桃苷對照品(批號:111521-201809,純度:94.9 %),葛根素對照品(批號:110752-201816,純度:95.4 %),芍藥苷對照品(批號:110736-201943,純度:95.1 %,中國食品藥品檢定研究院);甲醇,乙腈均為色譜純(美國默克公司),其他試劑均為分析純,水為超純水。
2 方法與結果
2.1 色譜條件[12-14]
色譜柱:Waters Acquity UPLC BEH C18柱(2.1 mm×150 mm,1.8 μm);流動相:乙腈(A)-0.5 %醋酸水溶液(B),梯度洗脫(0~20 min,8 %→25 %A;20~25 min,25 %→38 %A;25~31 min,38 %→65 %A;31~35 min,65 %→8 %A;流速:0.2 ml/min,檢測波長:230 nm(0~12 min),250 nm(12~17 min),330 nm(17~20 min),360 nm(20~23 min),284 nm(23~35 min);柱溫:30 ℃;進樣量:2 μl;色譜記錄時間:35 min。
2.2 溶液配制
2.2.1 混合對照品溶液的制備 分別取橙黃決明素、大黃酚、毛蕊花糖苷、京尼平苷酸、金絲桃苷、葛根素、芍藥苷對照品各適量,精密稱定,加甲醇制成每1 ml含橙黃決明素100.2 μg,大黃酚77.53 μg,毛蕊花糖苷21.67 μg,京尼平苷酸62.99 μg,金絲桃苷55.97 μg,葛根素40.08 μg,芍藥苷22.16 μg的混合對照品溶液,室溫避光保存。
2.2.2 供試品溶液的制備 取障眼明膠囊10粒,研勻,取粉末約0.5 g,精密稱定,置棕色具塞錐形瓶中,加稀鹽酸10 ml及三氯甲烷40 ml,加熱回流1.5 h,冷卻,分取三氯甲烷液,蒸干,殘渣加甲醇溶解并轉移至20 ml棕色量瓶中,加甲醇至刻度,搖勻,10 000 r/min離心10 min,取上清,0.22 μm微孔濾膜濾過,即得,室溫避光保存。
2.2.3 陰性對照溶液的制備 按障眼明膠囊制備工藝,分別制備缺決明子、車前子、菟絲子、葛根、白芍的陰性樣品,按2.2.2項下方法操作,制備陰性對照溶液。
2.3 方法學考察
2.3.1 系統適用性和專屬性試驗 分別取混合對照品溶液、供試品溶液及2.2.3項下的陰性對照溶液,按2.1項下色譜條件進樣測定,記錄色譜圖,見圖1。結果顯示,混合對照品溶液與供試品溶液中各指標成分色譜峰的理論塔板數均大于8400,分離度均大于1.5,拖尾因子在0.95~1.04之間,表明系統適用性良好。在樣品色譜圖中,分別有與對照品保留時間一致的特征峰,表明各化合物之間分離度好,無干擾,專屬性良好。
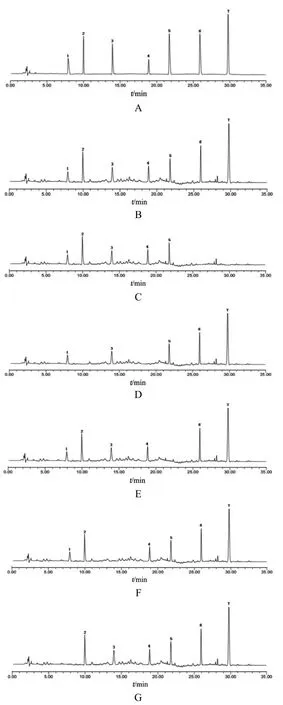
圖1 UPLC圖譜
2.3.2 線性關系考察 分別精密吸取2.2.1項下混合對照品溶液1,2,3,4,5,6 ml,置于同一25 ml棕色量瓶中,加甲醇溶液稀釋至刻度,搖勻,得系列濃度的混合對照品溶液,按2.1項下色譜條件測定,記錄色譜圖。以峰面積為縱坐標(Y),相應對照品濃度為橫坐標(X,μg/ml)進行線性回歸,繪制各組分標準曲線,計算回歸方程和相關系數。結果表明:橙黃決明素、大黃酚、毛蕊花糖苷、京尼平苷酸、金絲桃苷、葛根素、芍藥苷在試驗范圍內成良好的線性關系。精密量取2.2.1項下混合對照品溶液,用甲醇倍比稀釋,按2.1項下液相色譜條件進樣分析,峰面積信噪比10:1(S/N=10)時的混合對照品溶液濃度為定量下限(LOQ),峰面積信噪比3:1(S/N=3)時的混合對照品溶液濃度為檢測下限(LOD),結果見表1。
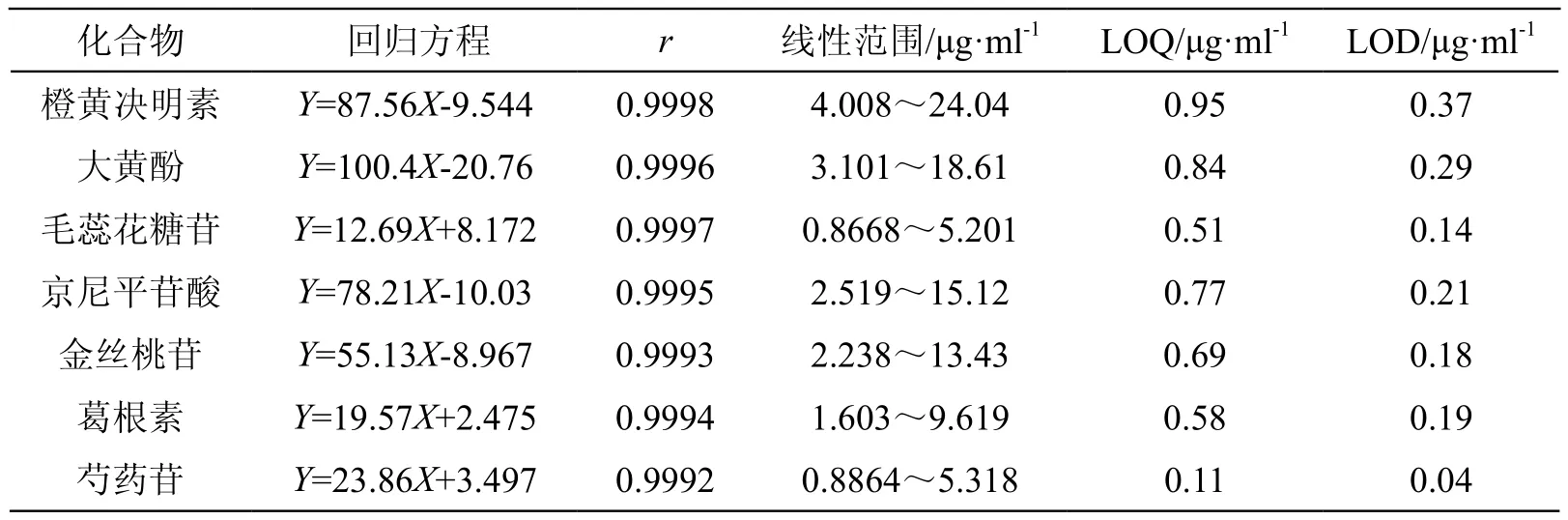
表1 分析物的回歸方程、檢測下限及定量下限
2.3.3 儀器精密度試驗 精密吸取2.2.1項下混合對照品溶液2.0 ml,置于同一20 ml棕色量瓶中,加甲醇溶液稀釋至刻度,按2.1項下色譜條件連續進樣6次,記錄峰面積。結果橙黃決明素、大黃酚、毛蕊花糖苷、京尼平苷酸、金絲桃苷、葛根素、芍藥苷峰面積RSD分別為0.55 %,0.67 %,0.46 %,1.01 %,0.71 %,0.61 %,0.82 %(n=6),表明儀器精密度良好。
2.3.4 穩定性試驗 取同一供試品溶液(批號:20191219),分別于制備后0,4,10,16,18,20,24 h,按2.1項下色譜條件進樣測定,記錄峰面積。結果橙黃決明素、大黃酚、毛蕊花糖苷、京尼平苷酸、金絲桃苷、葛根素、芍藥苷峰面積RSD分別為0.48 %,0.52 %,0.63 %,0.56 %,0.71 %,0.78 %,0.85 %(n=7),表明供試品溶液24 h內穩定性良好。
2.3.5 重復性試驗 取同一批供試品細粉約0.5 g(批號:20191219),精密稱定,共6份,按2.2.2項下方法制備供試品溶液,按2.1項下色譜條件進樣測定,記錄色譜圖,并計算7個成分的含量及其RSD值。結果橙黃決明素、大黃酚、毛蕊花糖苷、京尼平苷酸、金絲桃苷、葛根素、芍藥苷的平均含量分別0.6132,0.3866,0.0863,0.1333,0.0922,0.3074,0.1025 mg/g,RSD分別為0.71 %,0.59 %,0.92 %,0.63 %,0.83 %,1.08 %,0.76 %(n=6),表明方法重復性良好。
2.3.6 加樣回收率試驗 取已知含量的障眼明膠囊(批號:20191219)細粉6份,每份約0.25 g,精密稱定,精密加入橙黃決明素、大黃酚、毛蕊花糖苷、京尼平苷酸、金絲桃苷、葛根素、芍藥苷混合對照品溶液(質量濃度分別為77.25,48.75,10.92,16.86,11.75,38.85,12.95 μg/ml)2.00 ml,按2.2.2項下供試品溶液方法制備,按2.1項下色譜條件進樣測定,計算各成分加樣回收率及其RSD值。結果橙黃決明素、大黃酚、毛蕊花糖苷、京尼平苷酸、金絲桃苷、葛根素、芍藥苷的平均加樣回收率分別為99.73 %,99.66 %,99.56 %,99.03 %,99.12 %,99.02 %,99.59 %,RSD分別為1.01 %,0.71 %,0.92 %,0.65 %,0.70 %,0.76 %,1.08 %(n=6),表明本方法回收率良好,見表2。

表2 7個成分的加樣回收試驗結果(n=6)
2.3.7 樣品含量測定 按2.1項下色譜條件,分別精密吸取2.2項下混合對照品溶液和供試品溶液各2 μl,進樣測定,記錄橙黃決明素、大黃酚、毛蕊花糖苷、京尼平苷酸、金絲桃苷、葛根素、芍藥苷的峰面積,對4批障眼明膠囊中7個成分含量進行測定,結果見表3。
3 討論
本研究對供試品溶液進行190~400 nm全波長掃描,結果發現230,250,330,360 nm及284 nm波長下色譜峰豐度、分離度較好,故選擇利用PDA檢測器,采用波長切換法檢測橙黃決明素,大黃酚,毛蕊花糖苷,京尼平苷酸,金絲桃苷,葛根素,芍藥苷。依次考察了不同比例和梯度的流動相系統(甲醇、乙腈、0.5 %醋酸、0.5 %磷酸)、柱溫(20,25,30,35 ℃)、體積流量(0.1,0.2,0.3,0.4 ml/min)、進樣量(1,2,3μl)等對色譜峰的影響,以峰分離度、峰對稱因子等為依據進行了篩選,并分別優化了不同體積比的稀鹽酸和三氯甲烷提取溶劑(10:30,10:40,10:50)、提取方式(浸漬過夜、超聲處理、加熱回流)、定容溶劑(50 %甲醇、75 %甲醇、甲醇、乙醇、水)、提取時間(1,1.5,2 h),最終確定了2.1項下色譜條件及2.2.2項下供試品溶液的提取方法。
考察了Waters Acquity UPLC BEH C18(150 mm×2.1 mm,1.8 μm)色譜柱、Phenomenex Kinetex XB C18(150 mm×2.1 mm,1.8 μm)及YMC-Triart UPLC C18(150 mm×2.1 mm,1.8 μm)色譜柱的分離效能。結果顯示,各成分均能有效分離,但Waters色譜柱中各色譜峰峰形較好且對稱性滿足要求,因此選用Waters Acquity UPLC BEH C18(150 mm×2.1 mm,1.8 μm)色譜柱。
UPLC在分析效率、靈敏度和峰容量方面優于常規HPLC,并可顯著減少分析時間,可在短時間內達到平衡,同時相應會減少溶劑消耗[15-16],因此,已越來越多地應用于中藥分析研究。
本試驗采用1.8 μm小顆粒填料色譜柱,相比于HPLC使用的常規色譜柱,這一基于小粒徑的分析法具有更高的效率、更佳的數據質量,能顯著縮短分析時間,在保證所測定有效成分含量結果一致的情況下,分析時間節省了近30 min。
綜上,本試驗采用UPLC對障眼明膠囊中7個活性成分同時進行測定,方法操作簡便,精密度、穩定性、重復性好,對科學全面評價障眼明膠囊質量具有參考價值。
