基于紫精的陽離子型有機聚合物對鉬锝分離的應用研究
2021-12-27 11:05:16趙鵬偉陳善勇金永東夏傳琴
同位素 2021年6期
趙鵬偉,丁 沐,陳善勇,金永東,夏傳琴
(四川大學 化學學院,成都 610064)
99mTc配合物廣泛用于各種疾病診斷,全世界每年約有4 000萬次患者使用99mTc藥物進行臨床檢查[1-2]。99mTc主要通過99Mo/99mTc發生器產生,在發生器中,采用氧化鋁作為吸附99Mo的柱填料,99Mo經過衰變變成99mTc,使用NaCl溶液對99mTc進行洗脫。99Mo以235U為原料,通過核反應堆生產。該方法235U利用率低、放射性三廢多、成本高,同時面臨核擴散的風險。更嚴重的是,國際上生產母體核素99Mo的大多數反應堆面臨停堆檢修、關停或退役問題,99mTc供應存在短缺甚至中斷的風險,急需發展新的99mTc制備方法[3-4]。如基于98Mo(n,γ)99Mo反應的中子輻照法以及基于100Mo(γ,n)99Mo反應的加速器生產方法,這些方法不產生裂變廢物,降低環境污染風險,排除了高濃縮235U擴散危險,成為了目前99mTc制備研究的熱點[5-9]。但是這些方法面臨共同的問題,所得99Mo含有大量Mo同位素載體,比活度較裂變99Mo大為降低,經典的氧化鋁色譜柱發生器難以得到符合要求的99mTc洗脫液[6,10-14]。如何高效、安全地進行鉬锝分離成為該方法發展的關鍵。目前較為有效的柱色層分離材料有Dowex-1×8陰離子交換樹脂、ABEC樹脂以及活性炭材料。Dowex-1×8陰離子交換樹脂需要使用有機溶劑將材料上的99mTc淋洗下來,而有機溶劑容易殘留,不利于后續的臨床使用。而ABEC樹脂在堿性條件下可以從含大量Mo的溶液中吸附99mTc,然后在水溶液中解析出99mTc,但是所需溶劑種類多,操作繁雜,材料耐輻照性能差,制備的色譜柱一致性差等缺點妨礙了其在醫學領域中的應用[14-16]。
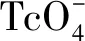
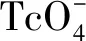
1 實驗材料與儀器
1.1 主要儀器
磁力攪拌器:C-MAG HS10,德國IKA集團;分析天平:AL204,瑞士METTLER TOLEDO公司;pH酸度計:pHS-3C,上海精密儀器有限公司雷磁儀器廠;旋轉蒸發儀:RE-2000B,上海亞榮生化儀器廠;水相針式濾器:聚醚砜SCAA-102,上海安普科學儀器有限公司;電感耦合等離子體發射光譜儀:Optima 8000,美國PerkinElmer公司;核磁共振:400 MHz,德國布魯克公司;傅里葉變換紅外光譜儀:Nicolet 670,美國尼高力儀器公司;X射線光電子能譜儀:XSAM 800,美國Kratos公司;X射線衍射儀:DX-1000,中國丹東方圓儀器有限公司;場發射掃描電鏡:S-4800,日本日立公司。
1.2 主要試劑
1,4,7,10-四氮雜環十二烷(>97%)、4-硝基溴化芐(99%):阿拉丁試劑有限公司;1-氯-2,4-二硝基苯(98%)、4,4′-聯吡啶(98%):薩恩化學技術(上海)有限公司;氯化亞錫、無水碳酸鉀、無水硫酸鈉,二水合鉬酸鈉:分析純,上海泰坦科技有限公司;乙腈、二氯甲烷、乙醚、鹽酸、甲醇、乙醇、1,4-二氧六環、氫氧化鈉、無水硫酸鈉:分析純,成都市科隆化工試劑廠;高錸酸鉀:分析純,東京化成工業株式會社。
2 實驗方法
2.1 有機合成
2.1.11,4,7,10-四(4-硝基苯基)-1,4,7,10-四氮雜環十二烷(TNBTA)的合成 參考文獻的合成方法[24],TNBTA的合成路線示于圖1。將7.52 g 4-硝基溴化芐(34.8 mmol)和4.81 g無水碳酸鉀混合在60 mL乙腈中,在室溫下向其中加入1.00 g四氮雜環十二烷(5.80 mmol)的乙腈溶液(60 mL)。在氮氣氛圍下室溫反應攪拌24 h。反應結束后過濾,濾渣用30 mL乙腈洗滌2次(2×30 mL),隨后用500 mL二氯甲烷溶解,高水洗滌5次,有機相用無水硫酸鈉干燥。減壓蒸去二氯甲烷后可以得到淡黃色固體,產率80%,核磁共振1H表征數據:1H NMR(400 MHz,CDCl3):δ 8.03(8H,d,1-H),7.45(8H,d,2-H),3.46(8H,s,3-H)2.64(16H,s,4-H)。
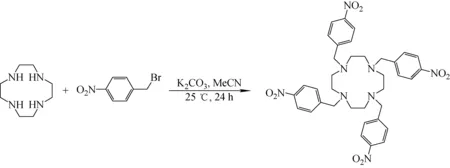
圖1 TNBTA的合成路線Fig.1 Synthesis route of TNBTA
2.1.21,4,7,10-四(4-氨基卞基)-1,4,7,10-四氮雜環十二烷(TABTA)的合成 參考文獻的合成方法[24],TABTA的合成路線示于圖2。將0.71 g TNBTA(1.0 mmol)加入到50 mL 70 ℃的濃鹽酸中,向混合溶液中加入3.79 g氯化亞錫(20.0 mmol)。在氮氣氛圍70 ℃下攪拌反應12 h,冷卻至0 ℃。過濾反應液,濾渣用少量高水溶解,向溶液中加入2 mol·L-1氫氧化鈉溶液,直至體系pH為13。混合液用二氯甲烷萃取3次,有機相用無水硫酸鈉干燥。旋蒸除去二氯甲烷后,得到產物,立即使用。TABTA的高分辨質譜和核磁共振1H譜表征數據如下。
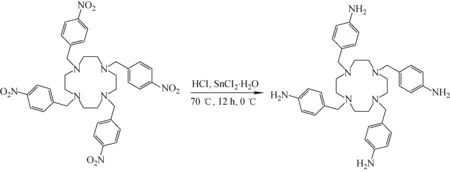
圖2 TABTA的合成路線Fig.2 Synthesis route of TABTA
1H NMR(400 MHz,CDCl3):δ7.11(8H,d,3-H),6.60(8H,d,2-H),3.58(8H,s,4-H),3.32(8H,s,1-H),2.63(16H,s,5-H)。
HRMS(ESI),m/zC36H49N8[M+H]+593.406 5;C36H48N8Na [M+Na]+615.381 7。
2.1.31,1′-雙(2,4-二硝基苯)-4,4′-二氯化聯吡啶(Zincke鹽)的合成 參考文獻的合成方法[24],Zincke鹽的合成路線示于圖3。將5.00 g 4,4′-聯吡啶(32.0 mmol)和22.68 g 1-氯-2,4-二硝基苯(112.0 mmol)溶解于175 mL無水乙腈中,混合溶液在氮氣氛圍下90 ℃攪拌回流72 h。反應結束后過濾反應液,濾渣分別用乙腈(60 mL)和乙醚(4×40 mL)洗滌。最終粉末真空干燥12 h,得到產物,產率20%。產物的高分辨質譜和核磁共振1H譜表征數據如下。

圖3 Zincke鹽的合成路線Fig.3 Synthesis route of Zincke salt
1H NMR(400 MHz,D2O):δ9.40(4H,d,4-H),9.33(2H,d,1-H),8.87(2H,d,2-H),8,85(4H,d,5-H),8.23(2H,d,3-H)。
HRMS(ESI),m/zC22H15N6O9[M-2Cl+OH]-507.086 4;C23H17N6O9[M-2Cl+CH3O]-521.100 8。
2.1.4材料VBCOP的制備 參考文獻的合成方法[24],VBCOP的合成路線示于圖4。將0.10 g新制備的TABTA(0.17 mmol)溶解于10 mL甲醇中,隨后加入到由0.19 g Zincke鹽(0.34 mmol)、10 mL甲醇和20 mL二氧六環構成的混合溶液中。反應溶液在氮氣氛圍下120 ℃攪拌回流72 h。反應結束后過濾,濾渣分別用乙醇、水、乙醚洗滌,最終粉末真空干燥12 h,得到產物,產率為53%。
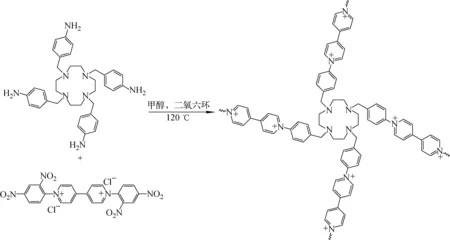
圖4 陽離子型有機多孔聚合物VBCOP的制備路線Fig.4 Synthesis route of VBCOP
2.2 靜態批次吸附
2.2.1pH對VBCOP吸附高錸酸根離子和鉬酸根離子的影響 固液比為0.25 g·L-1,將5 mg VBCOP加入到20 mL含有濃度為0.59 mmol·L-1的高錸酸鉀水溶液或濃度為0.37 mmol·L-1的鉬酸鈉水溶液中,pH為1~11,將所混合物恒溫攪拌所需時間后,使用0.22 μm聚醚砜針式過濾器過濾。水溶液中的錸或鉬濃度通過電感耦合等離子體發射光譜儀(ICP-OES)測定,三次測定結果取平均值。
2.2.2初始濃度對VBCOP吸附高錸酸根離子和鉬酸根離子的影響 固液比為0.25 g·L-1,將5 mg VBCOP加入到20 mL含有一定濃度高錸酸根(16.0~236.0 mg·L-1)或鉬酸根(16.7~200.7 mg·L-1)水溶液中,將所得混合物恒溫攪拌所需的時間后,使用0.22 μm聚醚砜針式過濾器過濾。水溶液中的錸或鉬濃度通過ICP-OES測定,三次測定結果取平均值。
2.2.3Mo/Re混合溶液對VBCOP吸附的影響 以98Mo(n,γ)99Mo生產99Mo為例,假設反應堆中子通量為1×1014n/cm2·s,熱中子俘獲截面為0.13 b,1 g天然Mo或富集為90%的98Mo輻照8 d后可產生約1.6 Ci99Mo,即比活度為每克98Mo中含1.6 Ci99Mo,99Mo經衰變后(忽略母體衰變損失),產生1.6 Ci99mTc[25]。使用與Tc物理化學性質相似的非放射性Re代替Tc,則Mo∶Re的質量比計算式如下,為1.75×106∶1。
摩爾比為:
固液比為1 g·L-1,將10 mg材料VBCOP加入到10 mL含有高錸酸鉀和鉬酸鈉的水溶液中,將所得混合物攪拌所需的時間后使用0.22 μm聚醚砜針式過濾器過濾,水溶液中錸以及鉬濃度通過ICP-OES測定。
3 結果與討論
3.1 結構表征

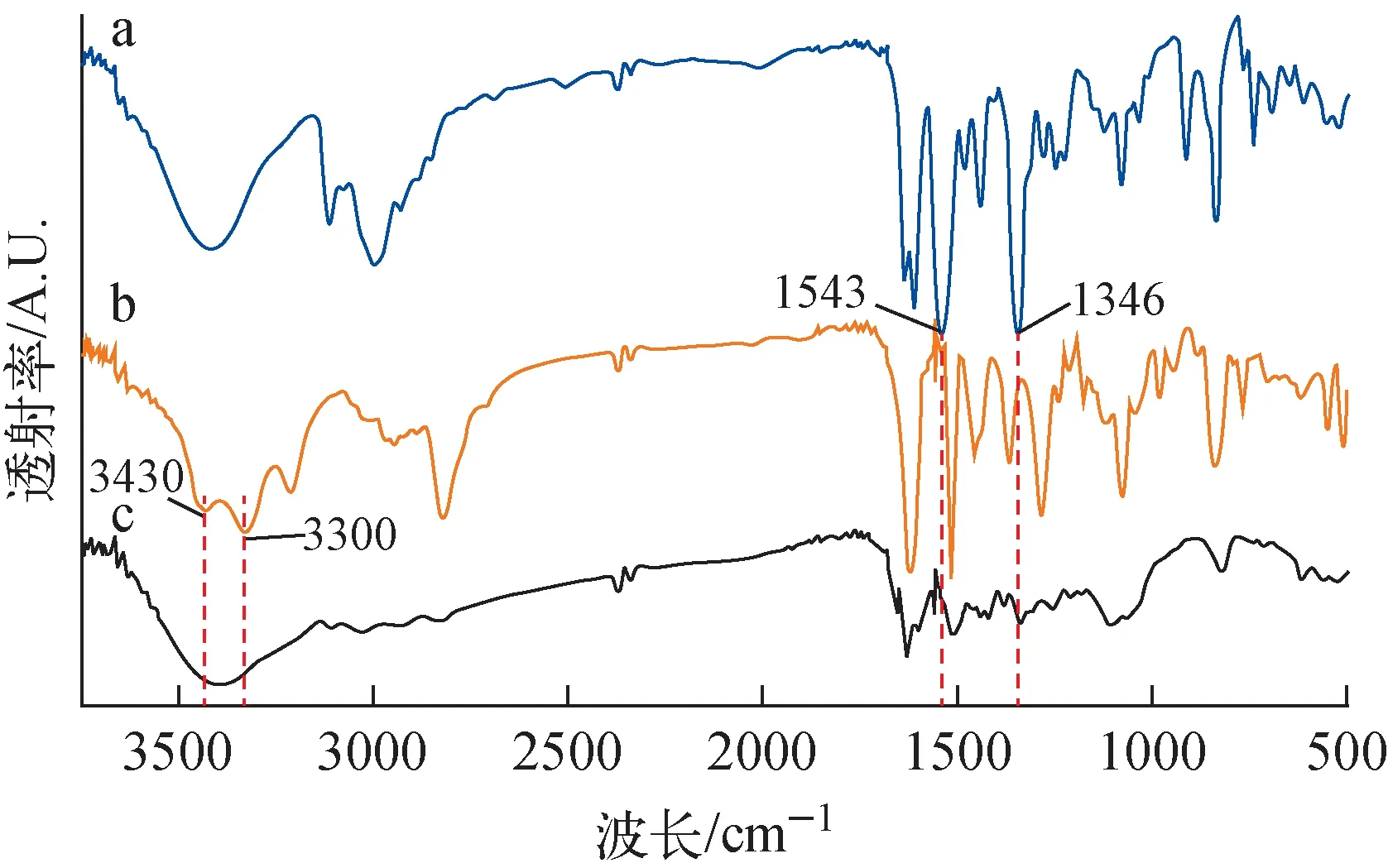
a——Zincke鹽;b——TABTA;c——VBCOP圖5 傅里葉變換紅外光譜Fig.5 Fourier transform INFRARED spectrum
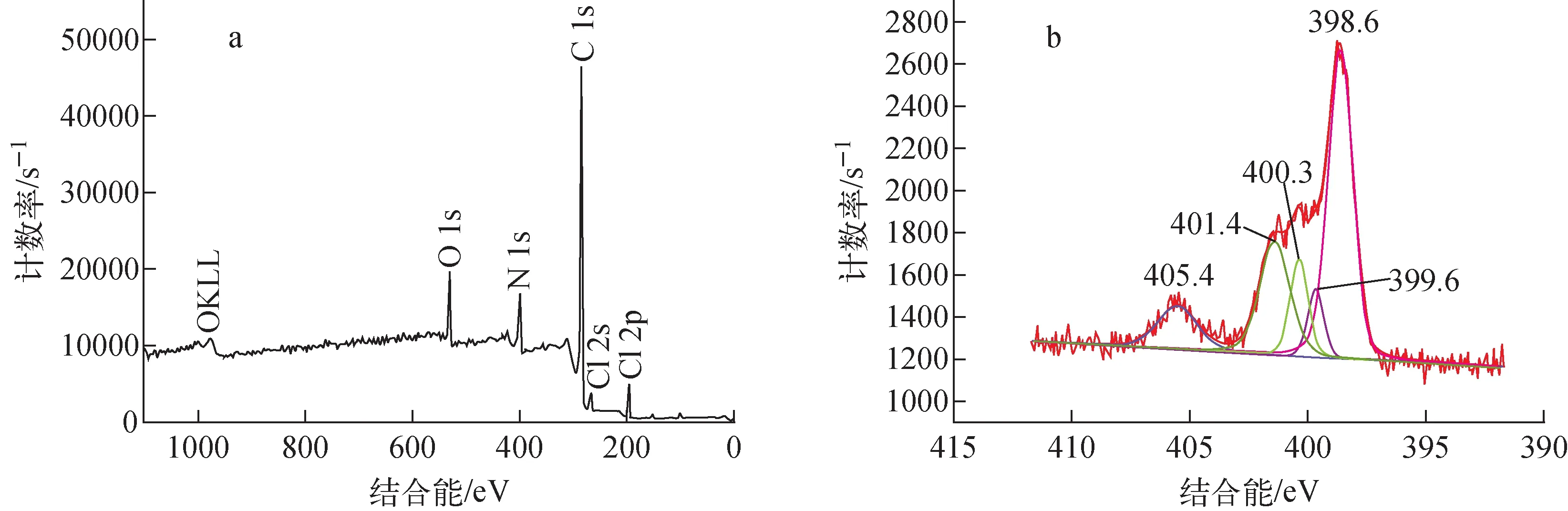
圖6 VBCOP的XPS全譜圖(a)和N 1s的分譜圖(b)Fig.6 XPS full spectrum of VBCOP (a)and N 1s XPS spectral fraction of VBCOP (b)
3.1.3粉末X射線衍射譜 粉末X射線衍射可以觀察材料的結晶情況。采用X射線衍射對制備的陽離子型共價聚合物進行表征,如圖7所示,制備的VBCOP材料在2θ角為15°~35°之間產生了一個較寬的衍射峰,表明該材料缺乏晶型,是無定型的材料。這主要是因為材料的構筑單元中存在柔性較高的四氮雜環十二烷結構。這種結構的存在不僅降低了材料單體之間的交聯度,使得材料網絡存在較多缺陷,而且使材料在分子層面缺乏剛性,不易于形成長程有序的晶相結構。
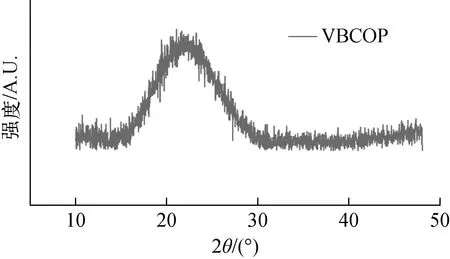
圖7 VBCOP的PXRD圖Fig.7 PXRD pattern of VBCOP
3.1.4掃描電子顯微鏡 制備的陽離子型共價聚合物材料的掃描電鏡圖像如圖8所示,VBCOP主要呈現光滑的球狀結構,直徑主要分布在2~6 μm。VBCOP的成球機理可以參考高分子聚合物的沉淀聚合法[31-32],即TABTA和Zincke鹽在溶液中發生Zincke反應,產生交聯度較低的低聚物,低聚物之間聚集合并而形成穩定的核進而從溶液中析出,形成的初級穩定核表面含有大量的反應位點,可以從溶液中捕獲TABTA、Zincke鹽及低聚物,進一步發生反應并不斷成長。這種機理從一定程度上也解釋了VBCOP具有無定型結構的原因。

圖8 VBCOP的SEM圖像Fig.8 SEM image of VBCOP
3.2 pH對VBCOP吸附高錸酸根離子和鉬酸根離子的影響
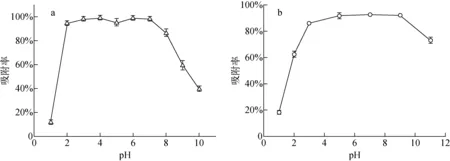
圖9 pH對VBCOP吸附和的影響Fig.9 Effect of pH on VBCOP adsorption of
3.3 初始濃度對VBCOP吸附高錸酸根離子和鉬酸根離子的影響
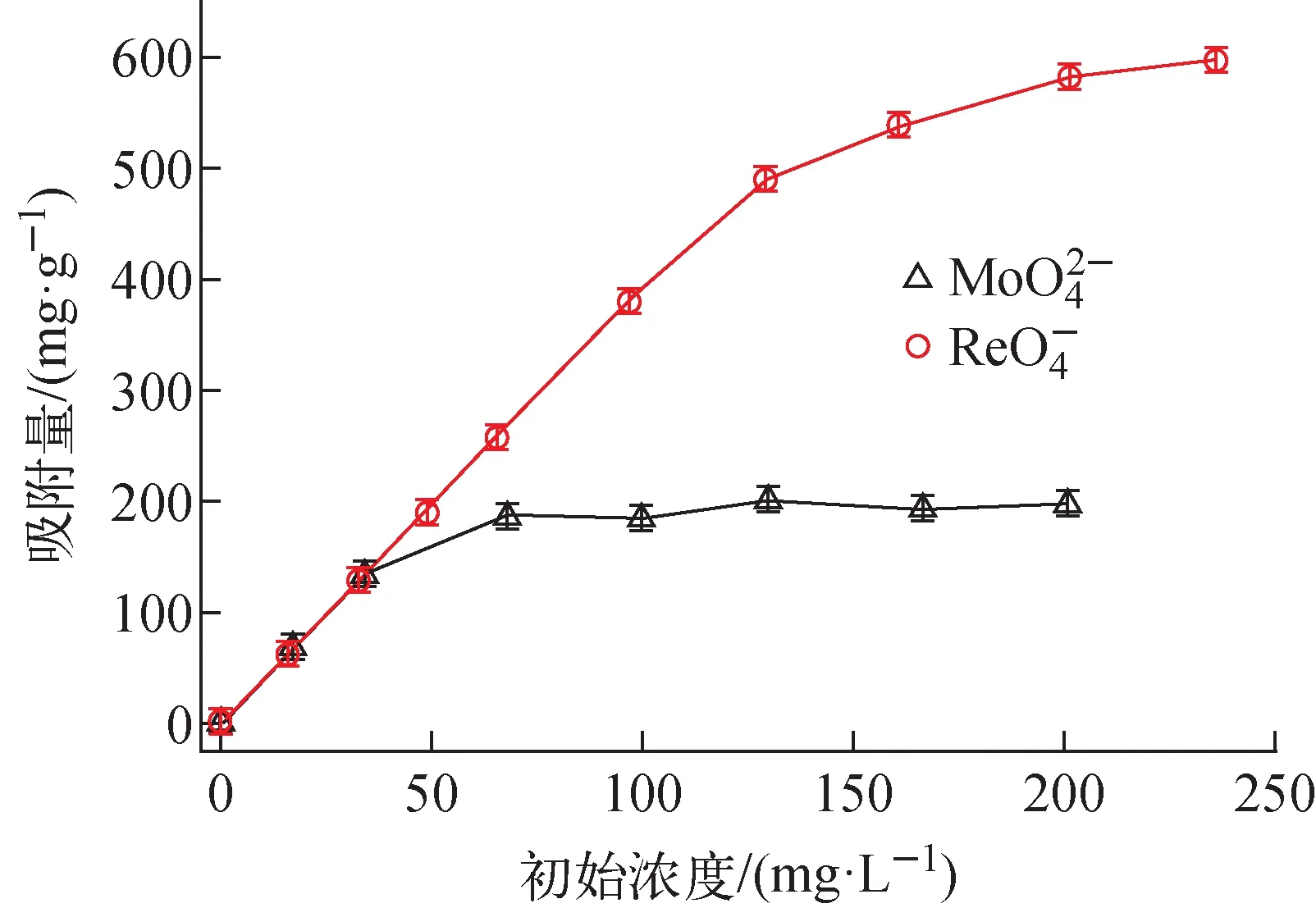
圖10 初始濃度對和吸附的影響(T=298 K,pH=7,m=5 mg,V=20 mL)Fig.10 Effect of initial concentration of
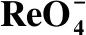
表1 VBCOP吸附與的等溫吸附模型擬合參數Table 1 Isothermal adsorption model fitting parameters for adsorption of by VBCOP
3.4 Mo/Re混合溶液對VBCOP吸附的影響
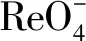
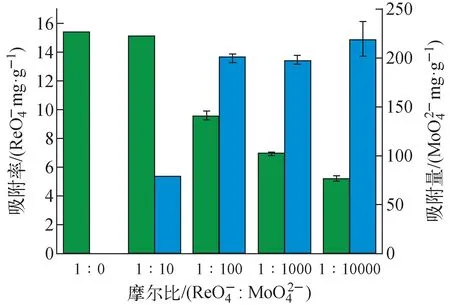
圖12 VBCOP在不同濃度比的情況下對(綠色)和(藍色)的吸附量(T=298 K,pH=7,m=5 mg,V=5 mL,Fig.12 The sorption capacity of at different concentration ratios
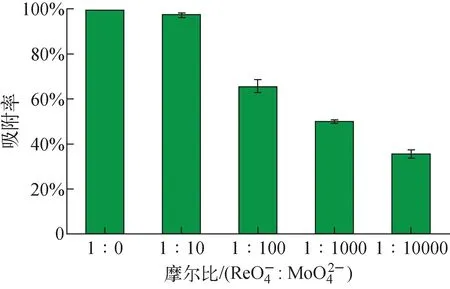
圖濃度對VBCOP吸附的影響(T=298 K,pH=7,m=5 mg,V=5 mL,Fig.11 Effect of concentration of on VBCOP adsorption of
4 結論
