ZnZr/HZSM-5雙功能催化劑在合成氣與苯烷基化反應中的催化性能
2021-12-28 01:24:20劉海華李艷春丁傳敏葛暉李學寬張瑋
化工進展 2021年12期
關鍵詞:催化劑
劉海華,李艷春,丁傳敏,葛暉,李學寬,張瑋
(1 太原理工大學化學化工學院,山西 太原 030024;2 中國科學院山西煤炭化學研究所,山西 太原 030001)
以甲苯、二甲苯為代表的輕質芳香族化合物是石化工業重要的基本有機原料之一,占據了石化工業領域三分之一的市場,特別是對二甲苯[1-3],被廣泛用于生產涂料、染料、樹脂、聚酯纖維和農藥等,由于其用途廣泛,需求量與日俱增。傳統的二甲苯主要來自石油餾分催化重整生成油和裂解汽油,但隨著石油和天然氣資源的嚴重短缺,通過石油路線得到的二甲苯產量遠不能滿足市場需求。
合成氣是一種以一氧化碳和氫氣為主要組分的混合氣體,作為碳資源轉化利用的樞紐,其來源非常廣泛[4],可從煤、天然氣和生物質等非石油類能源當中獲取,目前在我國市場廉價易得。合成氣作為眾多大型化工單元和石化過程的關鍵中間體,可以生產液體燃料、烯烴、芳烴以及醇類等下游產品。
合成氣與苯烷基化制甲苯和二甲苯等輕質芳烴是目前能源領域的研究熱點之一,可認為該技術是合成氣制甲醇和甲醇與苯烷基化的耦合[5-6],不僅可以充分利用市場上過剩的苯及合成氣資源,更重要的是為有效利用煤代替石油資源生產芳烴產物開辟了一條新興的工藝路線,具有很高的經濟效益,對于緩解芳烴資源短缺具有重要意義。
目前為止,研究者對合成氣與苯烷基化反應進行了大量的探索,如Zhao 等[7]采用浸漬法制備了Cu-Al2O3/ZSM-5催化劑并用于苯與合成氣烷基化反應,發現增加Cu 含量可以提高催化劑的活性,當Cu質量分數為11%時,苯轉化率可以達到16.80%,甲苯和二甲苯總選擇性達到80.00%。Yu等[8]在苯與合成氣烷基化反應研究過程中,將ZrO2與Zn浸漬的HZSM-5機械混合制成復合催化劑,實驗結果表明,HZSM-5中Zn質量分數為5%時,苯轉化率和二甲苯選擇性最高,分別達到31.00%和27.00%。Bai等[9]將ZnZr氧化物與ZSM-5機械混合,研究了Zn與Zr不同摩爾比和煅燒溫度對合成氣與苯烷基化性能的影響,發現當n(Zn)/n(Zr)=0.75,金屬組分煅燒溫度為500℃時,苯的轉化率為34.70%,甲苯和二甲苯總選擇性為86.80%。在合成氣與苯烷基化研究中,研究者采用各種方法對影響催化劑性能的因素進行了調變,在提高苯轉化率和目標芳烴選擇性方面已取得了較大進展,但仍然存在大多數催化劑活性偏低、穩定性不高等問題。
合成氣與苯烷基化反應過程復雜,產物眾多,但主要包含兩個步驟:首先,合成氣在金屬氧化物表面轉化生成甲醇或二甲醚;其次,甲醇或二甲醚遷移至分子篩Br?nsted酸位上與苯發生烷基化生成甲苯、二甲苯等。Zhao 等[7]以Cu/Al2O3/ZSM-5 為催化劑催化合成氣與苯烷基化,研究結果表明CO 首先在Cu/Al2O3上發生活化,并與氫氣反應生成二甲醚,二甲醚進一步在ZSM-5分子篩的Br?nsted酸位上與苯發生烷基化反應。Yang 等[10]在Pt/ZSM-5 催化劑上進行合成氣與苯烷基化反應,結果發現甲醇是合成氣與苯烷基化過程的中間體,中間體甲醇與苯在ZSM-5 分子篩的Br?nsted 酸和Lewis 酸共同催化作用下,生成目標產物甲苯和二甲苯。分析上述反應機理可知,合成氣與苯烷基化的高效催化劑至少包含兩種活性組分,甲醇合成活性組分和甲醇與苯烷基化活性組分。結合現有文獻報道可知,合成氣制甲醇主要在金屬氧化物上進行,而HZSM-5分子篩由于具有適宜的酸性,良好的水熱穩定性和優異的擇形性被廣泛應用于烷基化反應[11]。因此,將金屬氧化物與HZSM-5 分子篩耦合制備成同時具有合成甲醇活性組分和分子篩酸性催化功能的雙功能催化劑能夠高效催化合成氣與苯的烷基化。密度泛函理論(DFT)計算結果表明,合成氣在不同催化劑上制備甲醇的能壘分別為284.3kJ/mol(Rh/γ -Al2O3)[12]、 255kJ/mol (Mo6P3-Si3O9)[13]和177.6kJ/mol(Ni)[14],而甲醇與苯烷基化過程中甲氧基生成以及甲氧基與苯烷基化的能壘分別為149kJ/mol和97kJ/mol[15]。由此可知,合成氣與苯烷基化反應的決速步為中間體甲醇的生成,設計合成具有高CO 活化能力的金屬氧化物以快速地生成中間體甲醇是制備高效雙功能催化劑的關鍵。
大量研究結果表明,銅基催化劑是目前最主要的甲醇合成催化劑,具有操作溫度低、成本低和催化活性高等優勢[16],其在220℃下就能高選擇性地合成甲醇。但甲醇與苯烷基化過程需要的溫度一般為300~450℃,隨溫度升高,Cu基催化劑的主要產物逐漸變為甲烷,甲醇選擇性降低[17]。貴金屬催化劑同樣是重要的甲醇合成催化劑[18-19],如分子篩負載Pt 形成的復合催化劑,但是Pt 在沸石表面主要以單質Pt 和Pt2O 形式存在,高溫條件下會團聚形成較大顆粒,導致較低的催化穩定性,且貴金屬價格昂貴,實現工業化應用較為困難。研究表明,ZnO能夠促進H2的活化,ZrO2具有活化CO的能力,將二者結合可以催化合成氣制甲醇,而且可以通過改變二者的結合方式、摩爾比、焙燒溫度等條件對其催化性能進行調變[20],但是關于鋅鋯結合方式、含量對鋅鋯氧化物表面性質以及CO 活化能力影響的研究較少。
為此,本論文以鋅鋯金屬氧化物為生成中間體甲醇的活性組分,將其與HZSM-5分子篩耦合制備雙功能復合催化劑,并應用于合成氣與苯烷基化反應。ZnO和ZrO2相結合,一方面可以提高甲醇生成活性組分ZnO的分散度,另一方面能夠調變ZrO2表面的氧空位濃度,進而實現中間體甲醇的快速生成。為此,本論文系統考察了鋅鋯結合方式以及鋅含量對雙功能催化劑催化性能的影響規律,為合成氣與苯烷基化反應高效催化劑的制備提供一定的理論依據與指導。
1 實驗部分
1.1 催化劑制備
1.1.1 物理混合法
稱 取21.52g Zr(NO3)4·5H2O 溶 于100mL 去 離 子水中,配置濃度為1mol/L 的(NH4)2CO3溶液,當硝酸鋯溶液加熱至85℃時,緩慢滴加(NH4)2CO3溶液,直至溶液pH 為7 時停止滴加,老化2h,抽濾,100℃干燥,500℃焙燒4h得ZrO2。采用相同的方法以Zn(NO3)2·6H2O 制備ZnO。按n(Zr)∶n(Zn)=4∶1稱取ZrO2和ZnO并球磨5min得ZnO-ZrO2混合物。
1.1.2 浸漬法
稱 取3.02g Zn(NO3)2·6H2O 溶 于2mL 去 離 子 水中,待其完全溶解然后滴加到5g ZrO2中,60℃干燥12h,500℃焙燒4h 得到雙金屬氧化物,記為ZnO/ZrO2。
1.1.3 共沉淀法
稱取21.52g Zr(NO3)4·5H2O和3.71g Zn(NO3)2·6H2O溶于100mL 去離子水中,當混合溶液加熱至85℃時,緩慢滴加1mol/L(NH4)2CO3溶液,至溶液pH 為7 時停止滴加,老化2h,抽濾,100℃干燥,500℃焙燒4h得到雙金屬氧化物,記為ZnZryOx,y為鋯鋅摩爾比。
將上述不同方法制備的金屬氧化物與HZSM-5分子篩組合得到雙功能催化劑,如ZnZryOx-HZSM-5(a∶b)表示分段裝填,ZnZryOx/HZSM-5(a∶b)表示物理混合,其中(a∶b)表示氧化物與分子篩的質量比。
1.2 催化劑表征
XRD 譜圖在SHIMADZU-6000 X 射線粉末衍射儀上采集,Cu Kα射線(λ=0.154nm),2θ掃描范圍為5°~80°,掃描速度為5°/min;氧化物的形貌和元素分布在日本電子株式會社的JSM-7001F 型場發射電子顯微鏡(FE-SEM) 上進行分析;采用Thermo ESCALAB 250 型X 射線光譜儀微探針儀器(美國賽默飛)對氧化物表面物種進行分析;一氧化碳程序升溫脫附(CO-TPD)在TP-5076 型全自動化學吸附儀(天津先權)進行:稱取200mg氧化物置于400℃的He 氣流中預處理20min,降溫至35℃進行CO吸附,再繼續切換至He氣流吹掃樣品除去物理吸附的CO 后,以10℃/min 的升溫速率升至900℃,TCD檢測升溫過程中CO的脫附量。
1.3 催化反應和分析方法
將3.5g 催化劑(20~40 目)置于固定床不銹鋼管(內徑為10mm,長為750mm)中部,催化劑兩端裝滿石英球(10~20 目)并加熱至400℃。催化劑用氫氣處理3h 后將合成氣以2400h-1[GHSV(S)=2400h-1]的氣相空速通入反應器,至內壓達到3.5MPa,保持氣體空速不變,將苯以1.2h-1[GHSV(B)=1.2h-1]的空速經注射泵通入反應器內。
氣相和液相產物經冷阱分離后進行離線分析。氣相產物采用GC-920氣相色譜儀進行分析,配備了連接TCD 檢測器的5A 分子篩填充柱、碳分子篩填充柱和連接FID檢測器的改性氧化鋁填充柱。分析結果經CH4關聯,歸一化處理可得各產物的含量。液相產物采用GC-950 型氣相色譜儀進行分析,色譜柱為DM-Wax 毛細管柱,配備FID 檢測器,采用歸一化法對數據進行分析處理,得各產物含量。
催化劑活性指標有苯的轉化率(XB)和CO 的轉化率(XCO)。各產品收率指標分為兩類:一類與苯有關(SY),包括甲苯的選擇性(ST)、二甲苯的選擇性(SX)、乙苯的選擇性(SEB)、重芳烴的選擇性(SHA)和其他可忽略的產品;另一類與CO有關(SQi),包括芳烴側鏈的選擇性(SQ1)、C1~C5烷烴的選擇性(SQ2)、C2~C5烯烴的選擇性(SQ3)和CO2的選擇性(SQ4)。相關計算方法見式(1)~式(4)。

式中,Bin和Bout分別表示苯進料物質量和產物中苯的物質的量。

式中,COin和COout分別表示CO 進料物質的量和氣相產物中CO的物質的量。
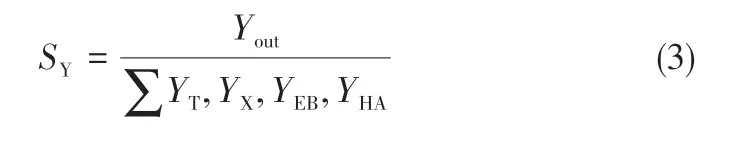
式中,Yout表示產物中目標芳烴的物質的量,包括甲苯(T)、二甲苯(X)、乙苯(EB)和重芳烴(HA)。

式中,Cisidechain表示產物中支鏈碳的物質的量,包括芳烴側鏈(Q1)、C1~C4烷烴以及C5非芳烴(Q2)、C2~C5烯烴(Q3)和CO2(Q4)。
2 結果與討論
2.1 催化劑表征
氧化鋯、氧化鋅及鋅鋯金屬氧化物的XRD衍射譜如圖1 所示。由圖1(a)可知,ZnO 為六角形的纖維礦相(PDF#76-0704);ZrO2為立方相(PDF#79-1769);ZnO-ZrO2樣品的衍射峰由純ZnO相和ZrO2相組成,說明兩相只是物理混合,沒有發生化學作用[21];ZnO/ZrO2樣品中ZnO 晶相的衍射峰很弱,表明ZnO 在ZrO2表面高度分散;ZnZr4Ox樣品形成了一種特殊的晶相結構,無ZnO和ZrO2晶相衍射峰,表明ZnO進入ZrO2晶格內部,二者形成了固溶體結構[22-23]。由圖1(b)可知,改變Zn 含量仍能保持固溶體結構,但是隨著Zn 含量的增大,出現微弱的ZnO的晶相衍射峰。
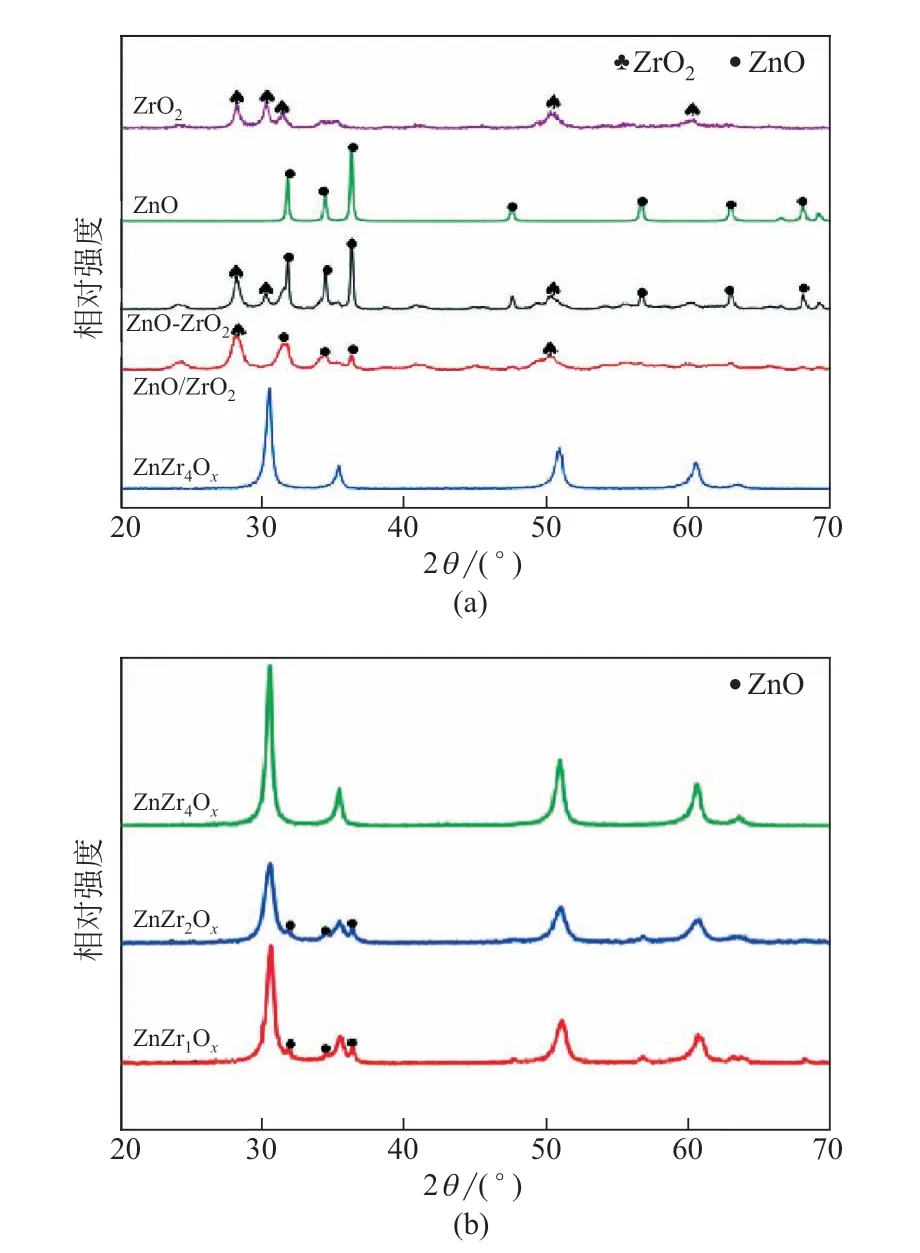
圖1 鋅鋯金屬氧化物的XRD圖
圖2所示的是不同方法制備的鋅鋯金屬氧化物的SEM 和EDS 圖。由圖可知,ZnO-ZrO2樣品中存在明顯的ZnO和ZrO2顆粒,進一步說明兩相只是物理混合,沒有發生化學作用。ZnO/ZrO2樣品中ZnO在ZrO2表面高度分散;ZnZr4Ox樣品中鋅鋯高度分散,形成一種均勻的結構。

圖2 鋅鋯金屬氧化物的SEM和EDS圖
由于鋅鋯金屬氧化物表面的氧空位是活化CO的活性位點[24],通過調變氧化物表面的電子結構進而增加表面氧空位含量是提高其催化活性的關鍵,因此采用XPS技術對鋅鋯金屬氧化物進行表征,結果如圖3 所示。圖3(a)為Zr 3d XPS 光譜,由圖可知,ZrO2、ZnO-ZrO2和ZnO/ZrO2樣品中Zr 的結合能相接近,表明浸漬法制備的樣品中鋅鋯之間存在的電子相互作用較弱。而ZnZr4Ox樣品中的Zr 向低結合能方向移動,主要由于Zr和Zn相結合,Zn最外層電子向Zr 移動,其電子云密度增加,電子結合能降低,表明固溶體結構中鋅鋯間存在較強的電子相互作用。圖3(b)為Zn 2p XPS 光譜,與Zr 3d XPS 光譜相類似,ZnO、ZnO-ZrO2和ZnO/ZrO2樣品中Zn的結合能相接近,但ZnZr4Ox樣品中的Zn向高結合能方向移動,進一步表明固溶體結構中鋅鋯間存在較強的電子相互作用[25]。圖3(c)為O1s XPS 光譜,結合能為528~531eV 的峰歸屬于晶格氧(Olattice),結合能為529~533eV的峰歸屬于缺陷氧化物或表面氧(Odefect)[26]。其中缺陷氧在總氧中所占比例如表1所示,由表可知,氧空位濃度按如下順序 遞 減 : ZnZr4Ox>ZnO/ZrO2>ZnO-ZrO2>ZrO2。ZnZr4Ox表現出最大的氧空位濃度,主要由于Zn 和Zr 之間形成固溶體結構,Zn 在ZrO2中高度分散且存在強的電子相互作用所致[27]。ZnO/ZrO2中Zn盡管在ZrO2表面高度分散,但二者之間相互作用力較弱,氧空位濃度增加幅度較小。而ZnO-ZrO2中氧空位濃度最低,主要由于ZnO和ZrO2只是簡單的物理混合,不存在電子相互作用[28-29],且ZnO 和ZrO2各自成相,所以氧空位濃度與ZrO2相接近。
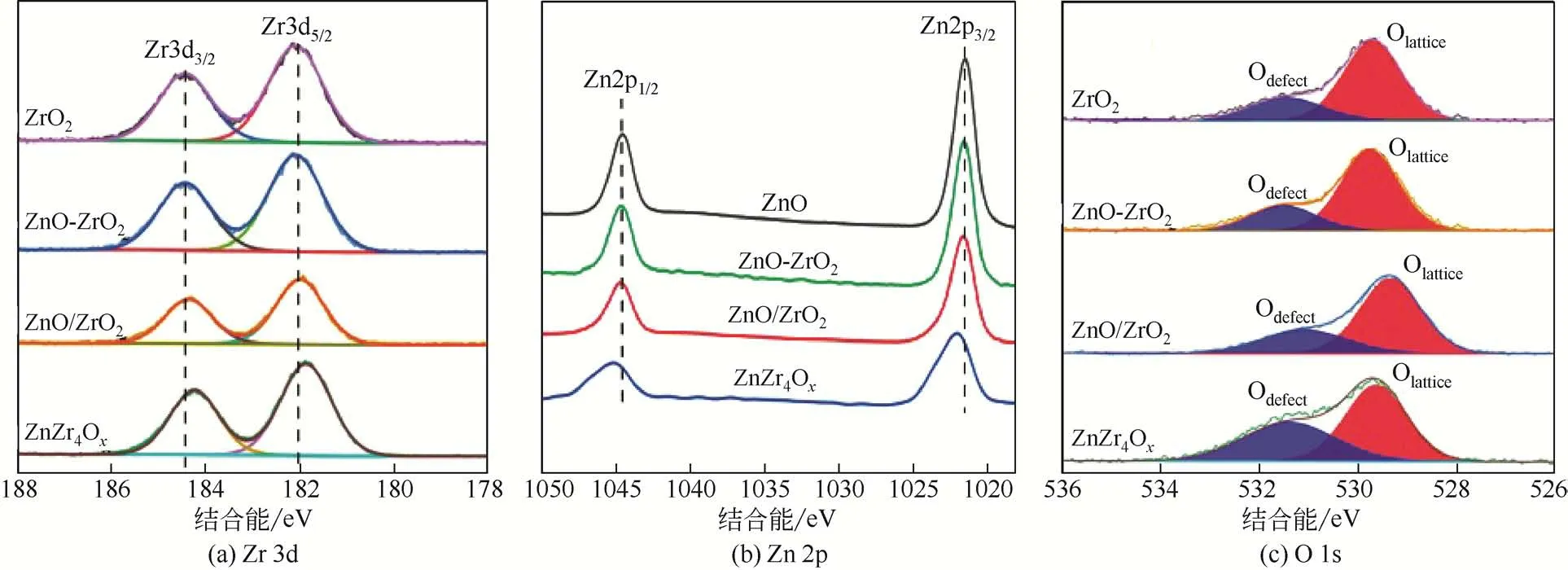
圖3 不同方法制備鋅鋯金屬氧化物的XPS圖
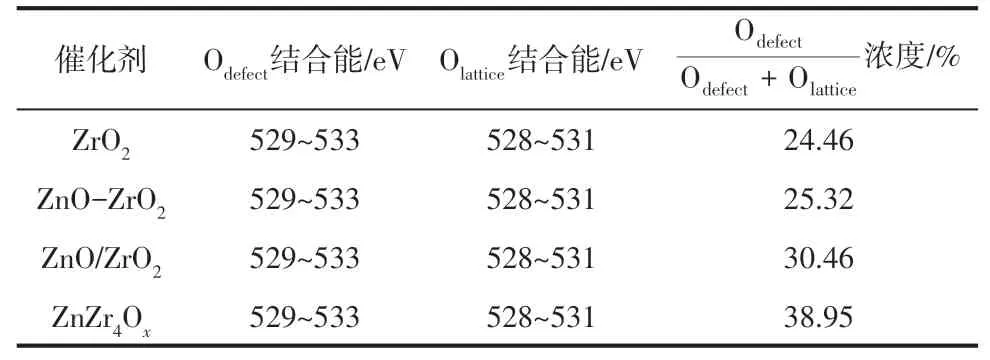
表1 鋅鋯金屬氧化物表面氧物種濃度
進一步采用CO-TPD對鋅鋯金屬氧化物的表面特性進行研究,結果如圖4所示,所有樣品出現兩個脫附峰,分別對應物理吸附(低溫)和化學吸附(高溫)。由圖4(a)可知,不同樣品對CO 的化學吸附量按如下順序逐漸遞減:ZnZr4Ox>ZnO/ZrO2>ZnO-Zr2O>ZrO2,與XPS表征的氧空位濃度相一致。ZrO2本身具有一定的氧空位,表現出一定的CO 吸附量,ZnO 的加入顯著地促進了其對CO 的吸附,其中ZnZr4Ox不僅表現出最大的CO吸附量,而且吸附能力也最強,主要由于Zn 和Zr 之間形成固溶體結構,Zn 在ZrO2中高度分散且存在強的電子相互作用,在其表面形成大量氧空位[28]。不同Zr/Zn 摩爾比的鋅鋯金屬氧化物CO-TPD 結果表明[圖4(b)],通過調變Zn的含量能夠改變鋅鋯金屬氧化物對CO的吸附量,主要由于Zn 能夠調變表面的氧空位濃度[30],當Zr/Zn摩爾比等于2時,鋅鋯金屬氧化物表現出最大的CO 吸附量,繼續增大Zn 含量,CO 吸附量不變,主要由于過量的Zn 物種以氧化鋅的形式存在。

圖4 鋅鋯金屬氧化物的CO-TPD
2.2 催化劑性能評價
2.2.1 鋅鋯不同結合方式對苯與合成氣烷基化性能的影響
雙功能催化劑中金屬氧化物的甲醇生成活性和分子篩的Br?nsted 酸性質[31-33]是影響合成氣與苯烷基化性能的主要因素,本文中所有雙功能催化劑采用相同的HZSM-5 分子篩,對反應活性的影響一致,因此催化性能的不同主要取決于雙功能催化劑中金屬氧化物的不同。
表2 所示的是不同鋅鋯金屬氧化物與HZSM-5耦合所得雙功能催化劑催化合成氣與苯烷基化反應18h 的反應物轉化率和產物選擇性。由表2 可知,ZrO2/HZSM-5 催化劑CO 的轉化率只有6.21%,而ZnO/HZSM-5 催化劑CO 的轉化率則高達16.06%,表明ZnO為合成甲醇的主要活性組分。與純ZnO相比,ZnO和ZrO2結合形成的鋅鋯金屬氧化物表現出更高的甲醇合成活性,結合方式不同,甲醇合成活性明顯不同,且按如下順序遞增:ZnO-ZrO2/HZSM-5<ZnO/ZrO2/HZSM-5<ZnZr4Ox/HZSM-5, 該順序與不同方法制備的鋅鋯金屬氧化物中Zn 的分散度以及表面氧空位濃度的變化順序相一致。隨著CO 轉化率的增加,苯的轉化率增加,氣相產物中烷基化選擇性增加,低碳烷烴的選擇性相應降低,表明苯與合成氣烷基化反應過程中甲醇的合成為決速步驟。另外,液相產物中二甲苯選擇性的變化規律與苯轉化率一致,與甲苯的變化規律剛好相反,表明合成氣與苯在雙功能催化劑上的反應順序為:金屬氧化物表面生成的甲醇在分子篩的B酸位與苯反應生成甲苯,甲苯進一步與甲醇反應生成二甲苯,符合連續反應機理。
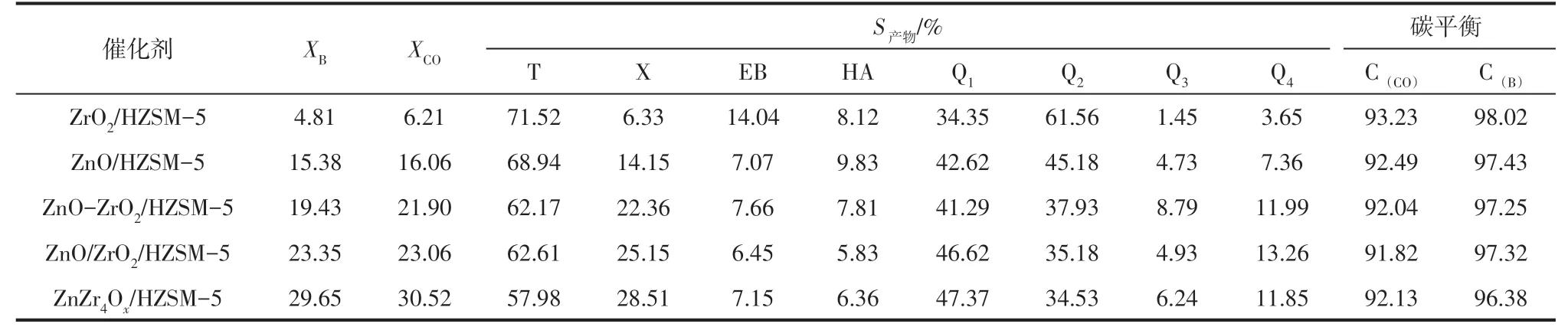
表2 不同雙功能催化劑催化合成氣與苯烷基化反應的反應物轉化率和產物分布
DFT 計算結果表明[12-15],合成氣與苯烷基化反應的決速步驟為甲醇的生成,因此反應的催化活性主要取決于金屬氧化物中的鋅含量以及表面氧空位濃度。金屬氧化物制備方法不同,雙功能催化劑的催化活性存在較大差異,主要由于不同方法制備的氧化物,其鋅物種的分散度和表面氧空位濃度不同。與純ZnO相比,后浸漬法制備的鋅鋯氧化物中Zn物種在ZrO2表面高度分散,使其催化活性增強,當采用共沉淀法將鋅鋯氧化物制備成固溶體時,不僅實現了Zn 物種的高度分散,且在其表面形成大量氧空位濃度,催化活性進一步提高。物理混合法制備的鋅鋯氧化物中ZnO和ZrO2各自成相,ZnO幾乎不分散,氧空位濃度與ZrO2相接近,所以催化活性相當于ZnO與ZrO2的線性加和。
雙功能催化劑催化合成氣與苯烷基化反應過程中反應物轉化率和產物選擇性隨時間的變化關系如圖5所示,由圖可以看出,所有催化劑在72h 內反應物轉化率和產物選擇性幾乎沒有變化,表明其具有很高的穩定性。
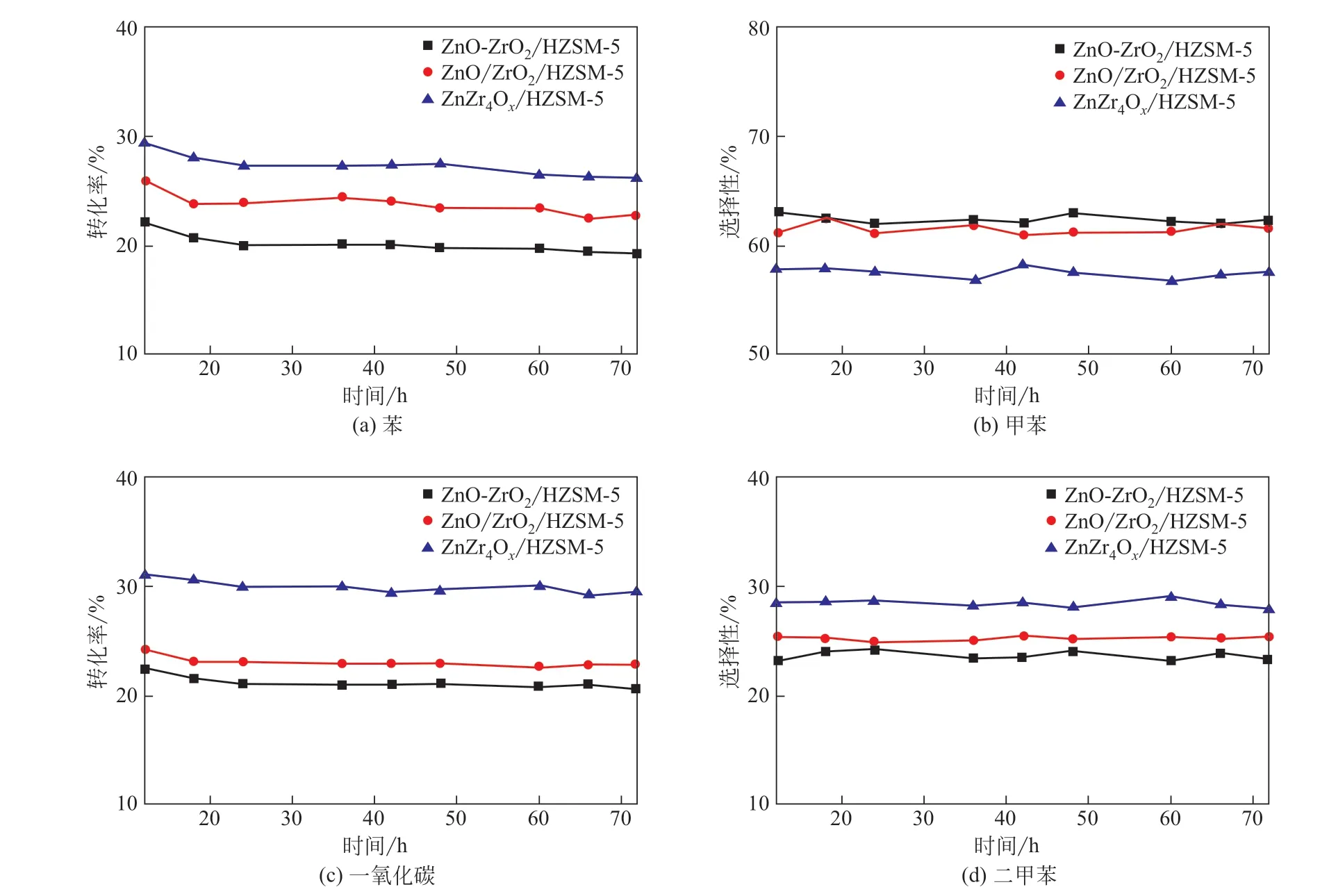
圖5 反應物轉化率和產物選擇性隨反應時間的變化關系圖
2.2.2 固溶體中鋅含量對苯與合成氣烷基化性能的影響
進一步考察了固溶體中Zn 含量對合成氣與苯烷基化反應性能的影響,結果如表3所示。由表可知,反應物轉化率隨Zn 含量的增加而增大,繼續增加Zn含量,反應物轉化率幾乎保持不變。
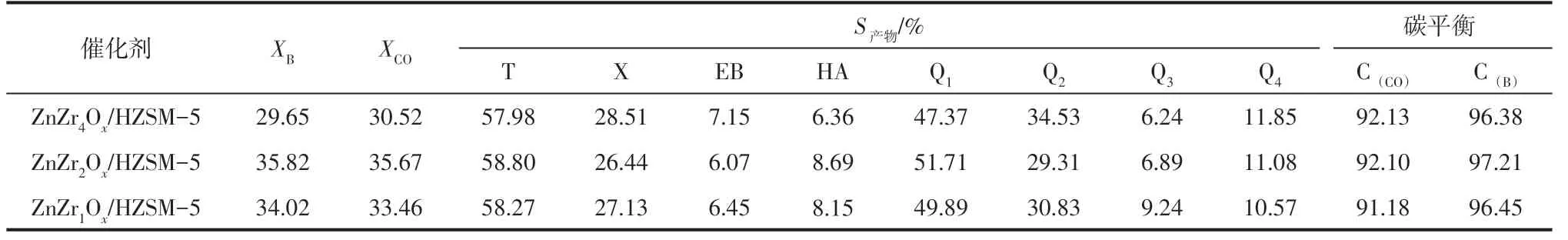
表3 不同Zn含量雙功能催化劑催化合成氣與苯烷基化的反應物轉化率和產物分布
采用共沉淀法制備的不同Zn 含量的鋅鋯金屬氧化物都具有固溶體結構,制備方法對反應性能的影響相同,因此對催化活性的影響主要源于固溶體中Zn含量的不同。鋅鋯固溶體中的Zn物種高度分散,因此催化活性隨Zn含量的增加而增大。此外,固溶體結構中氧空位濃度隨Zn 含量增加而增大,進一步促進催化活性的提高。當n(Zr)∶n(Zn)=2時,氧空位濃度達到最佳值,再繼續增加固溶體中Zn的含量,過量的Zn 物種以ZnO 的形式存在,氧空位濃度幾乎保持不變,反應活性相應保持不變。
2.2.3 鋅鋯氧化物與分子篩不同比例及裝填方式對苯與合成氣烷基化性能的影響
將金屬氧化物與分子篩按不同質量比物理混合制得雙功能催化劑,考察其二者所占比例對合成氣與苯烷基化性能的影響,在最優比例的基礎上進一步考察填裝方式對其性能的影響,結果如表4。
由表4 數據可知,當HZSM-5分子篩在雙功能催化劑中所占比例由1∶2 降低至1∶1 時,苯的轉化率降低,CO 的轉化率升高,主要由于雙功能催化劑中金屬氧化物所占比例增大,活化CO 的位點增多,相應的轉化率增大,而分子篩所占比例降低,B酸位點數減少,苯的轉化率降低,相應的烷基化選擇性降低,甲苯選擇性也降低,生成更多的低碳烴;當HZSM-5分子篩在雙功能催化劑中所占比例由1∶2 增加至1∶4時,苯和一氧化碳的轉化率都降低,主要由于雙功能催化劑中金屬氧化物的含量顯著降低,CO 活化位點減少,生成的甲醇中間體較少,苯的轉化率相應降低,但是大量的分子篩能夠為苯與甲苯烷基化提供足夠的酸性位點,因此表現出高的烷基化選擇性和甲苯選擇性,由于生成的甲醇量較少,限制了甲苯繼續甲基化,因此二甲苯選擇性較低。進一步鋅鋯氧化物與HZSM-5分子篩按最佳比例(1∶2)分段填裝,考察兩活性位之間距離對合成氣與苯烷基化性能的影響,結果發現將鋅鋯氧化物與HZSM-5分子篩分段裝填所得催化劑ZnZr2Ox-HZSM-5(1∶2)反應物轉化率顯著降低,主要由于金屬氧化物上反應得到的甲醇中間體不能及時轉移至分子篩B 酸位與苯發生烷基化反應,而是自身反應生成低碳烷烴和烯烴,因此表現出低的苯轉化率和烷基化選擇性。
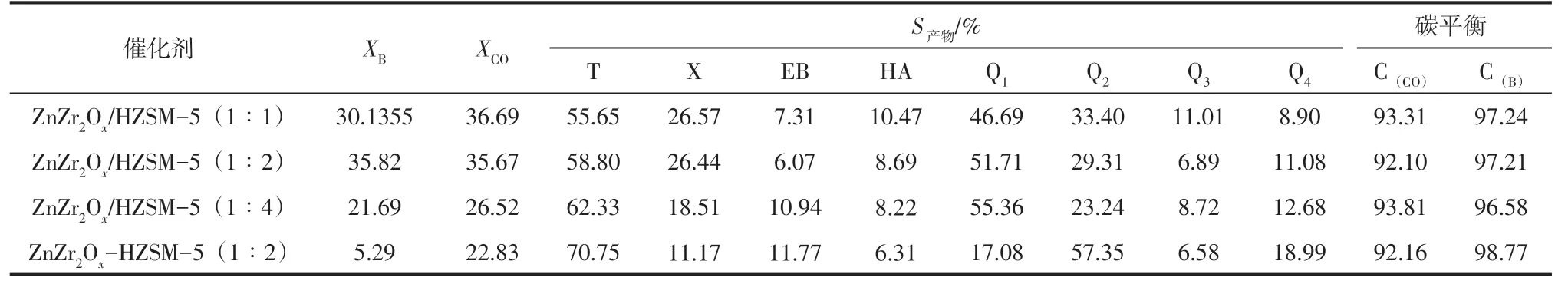
表4 不同比例及裝填方式的雙功能催化劑催化合成氣與苯烷基化的反應物轉化率和產物分布
3 結論
本文將鋅鋯金屬氧化物與HZSM-5分子篩耦合制備成雙功能催化劑,并對其催化合成氣與苯發生烷基化反應性能進行了研究,系統考察了鋅鋯金屬氧化物結合方式以及鋅含量對催化性能的影響,實驗結果表明ZnO 是合成氣制甲醇的主要活性組分,ZrO2表面的氧空位能夠活化CO 進而促進甲醇的生成,二者的結合方式會顯著影響Zn 物種的分散程度以及氧化物表面的氧空位濃度,進而影響催化活性,當鋅鋯形成固溶體時,Zn 的分散度最高,表面氧空位濃度最大,催化活性最高。隨著固溶體中Zn含量的增加,催化活性增大,當n(Zr)/n(Zn)=2的固溶體與HZSM-5 以質量比1∶2 耦合所得雙功能催化劑ZnZr2Ox/HZSM-5 表現出最高的催化活性,苯的轉化率為35.82%時,甲苯和二甲苯的總選擇性高達85.24%。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
