基于復雜網絡控制理論的腫瘤關鍵基因預測研究
2022-01-26 12:43:20詹秀秀張子柯
電子科技大學學報 2022年1期
關鍵詞:關鍵
姚 旭,詹秀秀,劉 闖*,張子柯,2
(1.杭州師范大學阿里巴巴復雜科學研究中心 杭州 311121;2.浙江大學媒體與國際文化學院 杭州 310018)
隨著人口的增長及老齡化,惡性腫瘤(即癌癥)已經成為人類死亡主要原因之一,是威脅生命健康的最大因素[1]。腫瘤癌變是環境因素和遺傳因素引起基因突變造成的,識別癌癥的致病基因對于精確腫瘤學至關重要,并且能夠促進靶向藥物的開發,對癌癥的治療具有指導意義[2-4]。
隨著第二代測序技術普及,以及人類基因組計劃、TCGA 計劃和ICGC 計劃的推進,研究者從大規模測序分析結果中明確了腫瘤存在著廣泛的異質性[5]。分布在不同患者個體中或者同一患者體內不同部位中的同種惡性腫瘤細胞,會產生從基因型到表型上的差異,相應地表現為多樣的基因突變[6]。高通量的測序數據使得科學家能從蛋白質水平中揭示腫瘤細胞發生機制,驗證腫瘤的相關基因突變,在癌癥中的應用有著廣泛的前景。文獻[7]證明了乳腺癌的蛋白質組學分析能夠解讀體細胞突變,縮小了缺失和擴增區域內驅動基因的候選提名范圍,并發現了相關治療靶標。文獻[8]從蛋白質組學研究入手,發現了白血病抑制因子(Leukemia inhibitory factor,LIF)是介導胰腺癌細胞和星狀細胞之間信號傳導的關鍵因子,并驗證了其可以作為胰腺癌治療的靶點和生物標志物。
然而,面對龐大的腫瘤數據,精準地找到腫瘤關鍵基因仍面臨著挑戰,僅從生物學角度研究腫瘤關鍵基因是遠遠不夠的。如今,癌癥這個復雜疾病又被稱為“網絡疾病”,可以從網絡的角度對生物學進行研究[9-10]。網絡中的節點可以代表生物分子,其相應的邊可以看作生物分子之間的功能、物理或化學相互作用[10]。因此,挖掘腫瘤的致病關鍵基因,找到潛在的控制疾病進展的靶點,可以從網絡控制入手。復雜網絡的控制理論和方法源于經典控制理論與復雜系統研究的結合,如果網絡中的一部分節點能夠在有限時間內將網絡從任意初始狀態變為任意期望的最終狀態,則該網絡稱為可控網絡[10-11]。文獻[12]在復雜網絡可控性方面做出了開拓性的研究,將網絡的可控性簡化為判定網絡結構可控性的問題,即忽略網絡中系統矩陣的邊權,只需關注系統內部的結構框架及節點間的連接方式。然而,文獻[12]提出的最大匹配算法只適合在有向網絡中尋找控制節點。為了應用于無向網絡,文獻[13]提出了最小控制集(minimum dominating set,MDS)方法,并發現MDS 方法在無標度網絡中只需要較小比例的節點就可以覆蓋控制網絡,并且網絡度分布的異質性越強,就越容易控制整個系統。
如今,復雜網絡控制理論已被廣泛應用到各種生物網絡分析中。文獻[14]在有向人類蛋白質相互作用網絡中應用最大匹配算法[12]確定了最小驅動蛋白集合,根據移除驅動節點后所包含的驅動節點數,對基因進行分類。此外,同時發現該可控性分析在疾病基因以及藥物靶點上的有關鍵作用。文獻[15]利用MDS 方法研究了蛋白質相互作用網絡的可控性,并確定了驅動蛋白集,分析表明該集合富含必需基因、癌癥相關基因以及病毒靶向基因,并且這些集合在網絡調控(富含轉錄因子和蛋白激酶)中有著重要作用。文獻[16]在MDS 模型的基礎上提出了一種中心性校正的最小支配集(centrality corrected-MDS,CC-MDS)模型,該模型比MDS 模型能夠捕獲更多的驅動蛋白。文獻[17]利用網絡的無標度特性算法[18]巧妙地避開了MDS 計算的復雜性,利用MDS各個解集中元素的角色,將節點分為3 類并應用于蛋白質相互作用網絡中,首次捕捉到了基因結構可控性、基因致死性和動態共表達的同步性之間的直接聯系。雖然網絡控制的方法可以捕捉生物網絡中的關鍵驅動蛋白及重要的相關調控功能,但是其對于各類癌癥中腫瘤關鍵基因識別預測方面還未得到分析驗證。
本研究利用復雜網絡控制理論的思想,從蛋白質相互作用網絡(protein protein interaction network,PPI)入手,通過對腫瘤發生的不同階段PPI 的變化進行分析,對腫瘤關鍵基因進行預測研究,可以闡明腫瘤蛋白表達水平的變化與腫瘤發生發展的相互關系及其規律。檢測、分析和確定腫瘤不同時期的標志蛋白,可以作為抗癌藥物篩選的作用靶點。該應用不僅對抗癌藥物發現具有指導意義,還可形成未來腫瘤診斷學、治療學的基礎理論。
1 研究方法
1.1 腫瘤關鍵基因預測分析流程
為了預測對腫瘤發展具有關鍵影響的基因,本文將復雜網絡控制理論應用于人類相互作用蛋白數據中,結合控制集和復雜網絡的拓撲性質,對蛋白質相互作用網絡中的基因進行了篩選,并對最終預測的腫瘤關鍵基因進行了有效的驗證。圖1 描述了腫瘤關鍵基因預測分析的流程,主要包含3 個步驟:1)通過最小控制集和控制分類模型將蛋白質網絡中的基因進行分類;2)對得到的各分類中的基因集合利用綜合中心性進行排序,進行富集分析,篩選出最終可用來預測的腫瘤關鍵基因;3)將得到的腫瘤關鍵基因,通過突變數據及相應的文獻分析,證明預測的腫瘤關鍵基因對腫瘤發展的影響力。Z_score 衡量的是觀測值與平均值間的差異,p指的是在假設檢驗中,當原假設為真時所得到的樣本觀察結果或更極端的結果出現的概率。
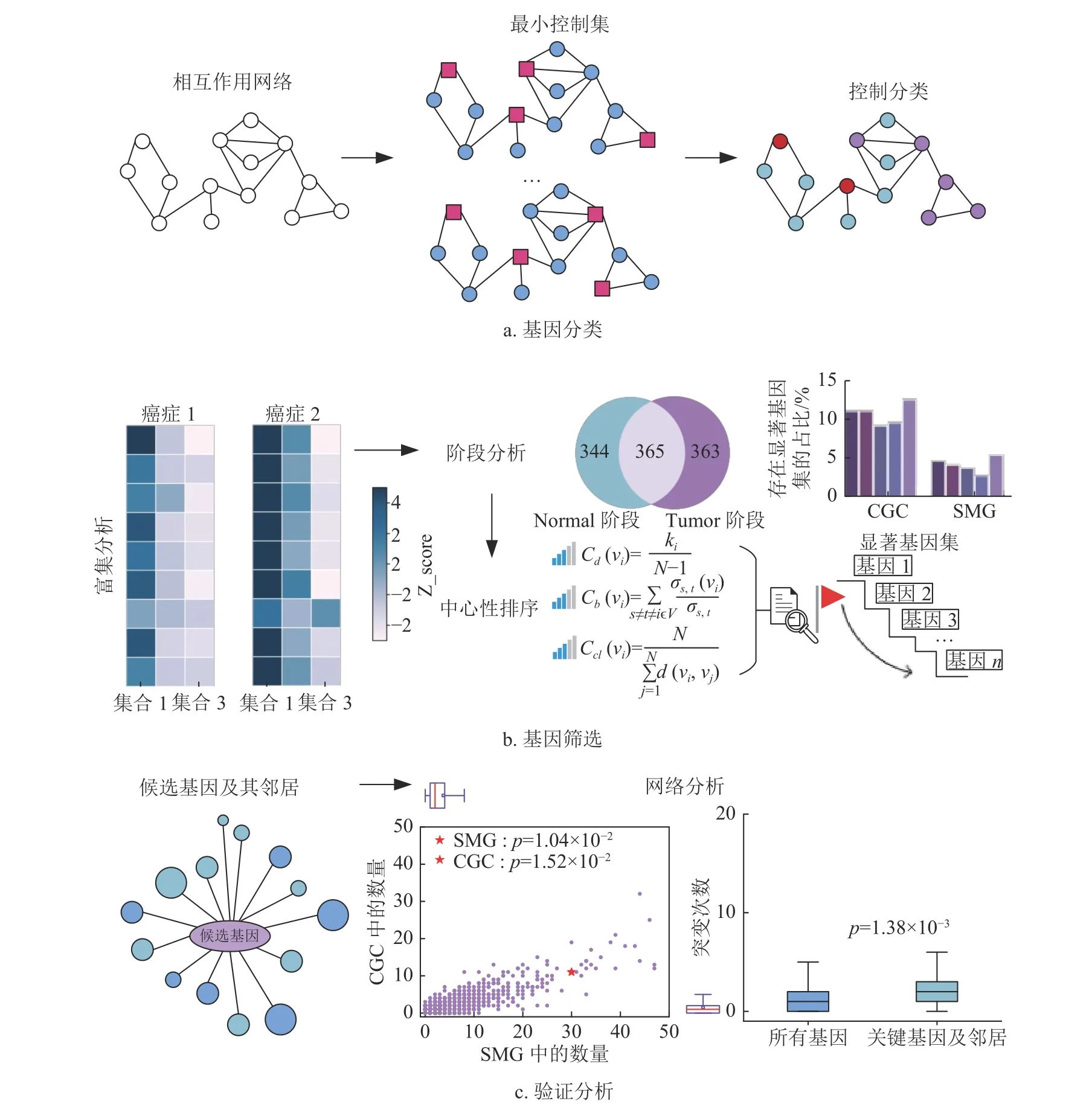
圖1 腫瘤關鍵基因預測分析流程
1.2 最大連通子圖
假設G=(V,E)表 示一個無向圖,其中V={v1,v2,···,vn}為G的 節點集,N為無向圖中節點的個數,E為G的 邊集。如果從頂點vi到 頂點vj中有路徑存在,則稱vi和vj連 通。存在子圖G′,如果其中的任意兩個頂點之間都連通并且為連通的子圖中最大的一個圖,則該圖G′為 無向圖G的最大連通子圖。
在蛋白質相互作用網絡中,節點代表蛋白質,邊代表兩個蛋白質之間的相互作用關系。其最大連通子圖就是蛋白質相互作用網絡中的最大的一個連通網絡,網絡中任意兩個蛋白質之間都能夠直接(或間接)相連通。
1.3 最小控制集模型
圖G=(V,E)中 存在一個節點集S?V,如果節點vi∈V,其要么是節點集S的一部分,要么與節點集S里一個元素相鄰,則稱這樣的節點集S為控制集(如圖1a 中間網絡中的紅色方形節點)。控制集S里的這些節點是圖1a 中的最優節點集,它對整個圖的結構有著支撐作用,除此之外的每個節點都能夠通過一條連邊與控制集S里的一個節點相連。本文定義在圖G的所有可能的控制集當中節點數最少的集合為MDS。
MDS 的概念屬于圖的控制理論。由于控制問題是經典的NP 完全問題,要解決這個未知能否在多項式時間內求解的問題,需要把它歸結為一個二進制的整數規劃問題(integer linear programming,ILP)來計算。本文通過尋找二進制向量x來最小化具有線性約束下的線性函數f(x)。圖G=(V,E)中每個節點vi的都有一個值為0 或1 的二進制整數變量xi,屬于控制集的節點的二進制變量取值為1,否則為0。于是,線性函數f(x)的約束表示為:
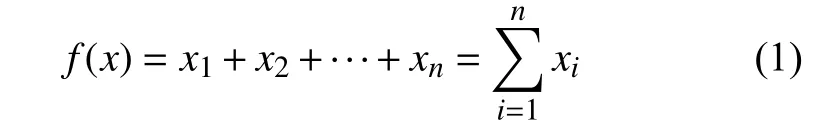
并且滿足約束:

式中,N(i)表 示節點vi的 鄰居;而n是圖中的節點總數。
最小控制集能夠提供對整個網絡起控制作用的節點集。蛋白質相互作用網絡中的這些最小控制集,能夠通過其相互作用影響整個PPI 網絡。解決最小控制集這個二進制整數規劃問題,可以利用分支限界算法來尋找這二進制線性函數約束問題的最優解。
1.4 控制分類模型
在一個連通的蛋白質相互作用網絡中,尋找控制基因集的最優解,往往會有多種符合最優解的解決方案。單純地將其中的一個最小控制基因集作用于復雜網絡中,并不能確定始終能夠影響整個網絡的一些關鍵節點的角色(即對整個癌癥網絡有確定影響的基因集),因此需要對集合進行進一步的分類。為了解決這個問題,文獻[13,17]根據每個節點在不同配置中參與的角色,提出了分類算法,構建了控制分類模型:始終存在于配置方案的關鍵節點(critical)、從不參與任何配置方案的冗余節點(redundant)、屬于某些配置方案但不存在于其他配置方案的間歇性節點(intermittent)。這3 類節點分別對應著圖1a 右側網絡的紅色、藍色和紫色的圓形節點。為了優化計算速度,文獻[13,17]在分類之前利用所證實的推論,對網絡中的節點進行了預處理,利用拓撲性質提前確定了一些節點的類型。
推論1 關鍵節點推論。如果節點vi有兩個或兩個以上的度為1 的鄰居結點,那么這個節點vi被定義為一個關鍵節點。
推論2 冗余節點推論。如果節點vi所有的鄰居結點都是關鍵節點,那么這個節點vi被定義為一個冗余節點。
本文整合了該控制分類模型,并將網絡中的節點進行了分類,算法流程如下所示。
算法1 節點的控制分類
輸入:節點集V、邊集E
輸出:關鍵節點集Gcritical、冗余節點集Gredundant、間歇性節點集Gintermittent


利用控制分類模型,將蛋白質相互作用網絡中的基因從復雜的相互作用關系中分類出3 部分,每部分集合中的基因之間對網絡控制的作用相同。
1.5 綜合中心性
文獻[19]提出的中心性-致命性規則指出,蛋白質相互作用網絡中高度連接的蛋白質往往是必不可少的。中心性是衡量網絡節點重要性的重要指標,能夠刻畫節點在網絡中的地位。如度中心性描述了一個節點對其他節點的直接影響;介數中心性刻畫了經過一個節點的最短路徑數,表明這個節點對網絡最短傳輸的控制力;接近中心性反映了一個節點與其他節點之間的接近程度,此節點可以通過鄰居節點能夠迅速地覆蓋到整個網絡。綜合中心性是通過加權的方法,將度中心性、介數中心性、接近中心性歸一化,并結合起來衡量節點在網絡中的綜合表現。為了精確地篩選腫瘤基因集中的更為關鍵的基因,本文利用綜合中心性來衡量基因對生物網絡的重要程度,并對從蛋白質相互作用網絡中分離出來的腫瘤基因集進行了排序。相關中心性的定義為:
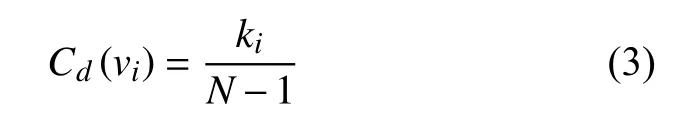
式中,Cd(vi)表 示節點vi的 度中心性;ki表示節點vi的 鄰居節點的數目;N表示節點個數;N?1指節點可能的最大度值。
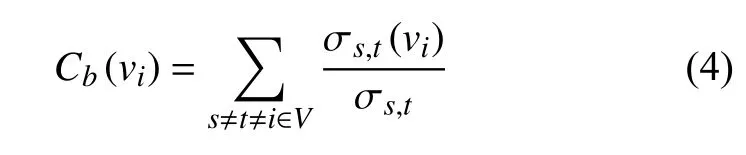
式中,Cb(vi) 表 示節點vi的介數中心性;σs,t表示節點s與節點t之 間最短路徑總數;σs,t(i)表 示節點s與節點t之間經過節點vi的最短路徑數目。
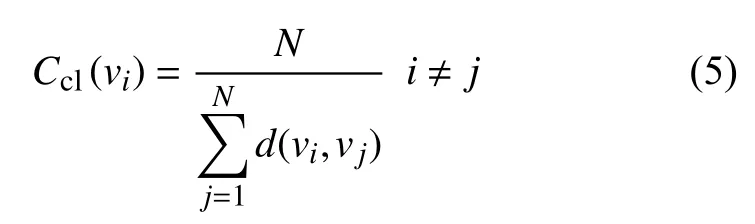
式中,Ccl(vi)表 示節點vi的 接近中心性;d(vi,vj)表示節點vi與節點vj的距離。
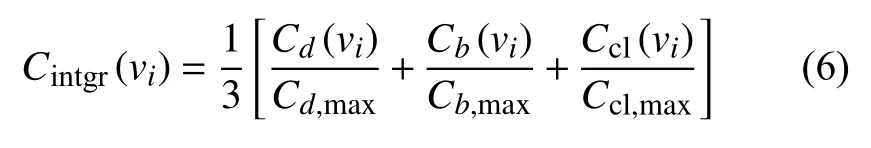
式中,Cintgr(vi)表 示節點vi的 綜合中心性;Cd,max,Cb,max,Ccl,max分 別為{Cd},{Cb},{Ccl}中的最大值。
2 數 據
2.1 蛋白質相互作用網絡
本文首先整合了多個人類蛋白-蛋白互作數據庫的PPI 數據(包含InnateDB[20]、PINA[21]、BioGrid[22]及HINT[23]等),刪除其中重復的邊和自環,構成了由15 474 個蛋白形成的170 631 條邊的網絡。本文從TCGA 數據庫[24]中收集了包括乳腺癌(BRCA)、腎透明細胞癌(KIRC)、肺腺癌(LUAD)、結腸腺癌(COAD)和頭頸鱗狀細胞癌(HNSC)在內的5 種癌癥類型及其相應的正常樣本的RNA-Seq (RPKM)數據,對于每個基因對,計算其共表達的皮爾森相關系數及相應的p值。將不同的癌癥類型的共表達關系嵌入到PPI 網絡中,選擇顯著的連邊(p<0.05)構成癌癥特異性蛋白-蛋白網絡[25]。
2.2 生物通路數據
本文從WiKiPathways 代謝通路數據庫[26]中收集了上述5 種癌癥類型的生物通路數據集,并利用一些已知與癌癥相關的生物通路,進一步得到每個與癌癥相關的生物通路的基因集,這些基因對各生物功能起著調控作用。
2.3 顯著基因集
本文收集了SMG (significantly mutated genes)[3],CGC (cancer gene census)[27]這兩個腫瘤基因集,這些基因集的基因在癌癥變化發展中所起到的重要作用都已被廣泛證實。
2.4 突變數據集
本文從TCGA 數據庫中下載了上述5 種癌癥類型的腫瘤病人的體細胞突變數據。
3 實驗與分析
3.1 基于網絡控制理論的基因分類
本文對TCGA 數據庫中的5 種癌癥類型的蛋白質相互作用數據進行了處理,從中提取出了具有顯著意義的連邊(即p<0.05),并利用最大連通子圖的定義,剔除了一些在網絡中最大連通子圖之外的小簇基因集,重構了蛋白質相互作用網絡,各個網絡的統征如表1 所示。
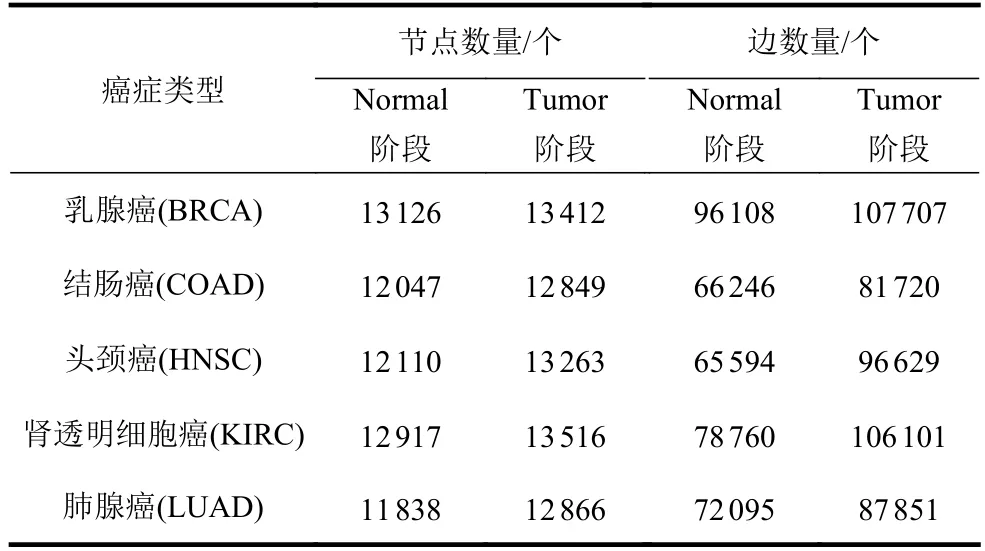
表1 腫瘤不同階段的最大連通子圖的節點連邊情況
為了從各個階段復雜的蛋白質相互作用對中,找到對腫瘤發展具有關鍵影響的基因集,本文利用復雜網絡控制理論中的最小控制集模型,并通過控制分類模型將各癌癥網絡各階段的基因進行了分類,研究這些基因對網絡產生的具體影響。通過方法1.3 和1.4,得出了各癌癥網絡各階段的3 種類型的基因集:關鍵(critical)基因集、間歇性(intermittent)基因集、冗余(redundant)基因集,其包含的基因情況如表2 所示。從最小控制集模型和控制分類模型的定義中,可以了解到關鍵基因集中的基因是滿足所有最小控制集配置方案的集合,即不管最小控制集的配置如何變化,這些基因始終包含在控制集中,其他非控制集中的基因都可以通過這些基因相互作用而到達。因此,關鍵基因集這一分類更有可能符合對腫瘤關鍵基因的研究預測集合的預期,于是本文提出了一個猜想:關鍵基因集中的基因能夠控制影響腫瘤的變化發展,在腫瘤突變中較為顯著。
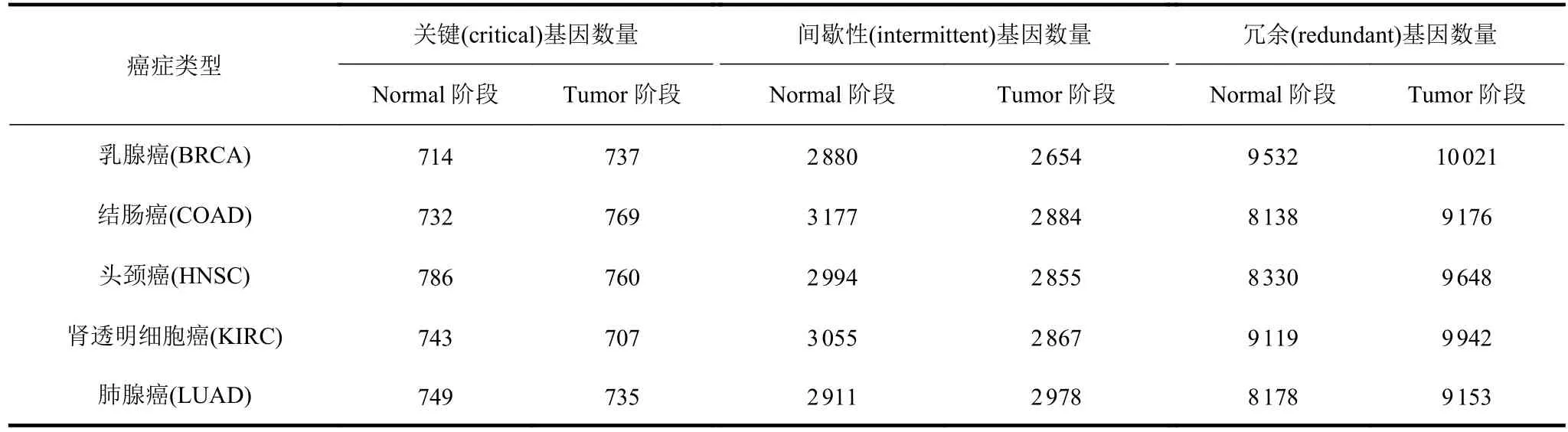
表2 各腫瘤不同階段的基因分類個數
3.2 腫瘤關鍵基因集的篩選
為了驗證本文的猜想,即關鍵基因集所包含的基因對癌癥發展的影響顯著明顯于其他兩種類型的基因集,本文將獲取的癌癥相關的生物通路數據集和顯著基因集與這3 種類型的基因集進行了富集分析。顯著基因集包含了CGC 基因集和SMG 基因集,它們所涉及的基因已被廣泛地被證實對癌癥具有重要影響;生物通路數據集中包含了多個與癌癥相關的生物功能作用,每個功能都有與其作用相關的基因組。結合并利用上述數據,分析3 種類型的基因集合在其中的富集情況,評估它們預測癌癥關鍵基因的性能。本文從網絡中隨機生成了10 000次與3 種類型基因集同等大小的集合作為參考,并與獲取的數據集做了交集,來衡量這3 種類型基因集相對于隨機基因集的顯著程度。本文利用了ZScore 值來表示3 種基因集的顯著性,圖2 顯示了腫瘤樣本Tumor 的3 種類別的基因集在共有的7 種生物通路功能和顯著基因集(SMG 和CGC)的富集程度(正常樣本Normal 的富集情況與其類似)。圖中橫坐標中C 表示critical;I 表示intermittent;R 表示redundant,縱坐標中AG 表示angiogenesis;CC 表示cell cycle;DDR 表示DNA damage response;ICP 表示integrated cancer pathway;MR 表示mismatch repair;SPIG 表示signaling pathways in glioblastoma;TN 表示TP53 network。從圖中可以看到:關鍵基因集具有統計學意義,富集度明顯優于間歇性基因集、冗余基因集。結果表明,關鍵基因集能夠很好地解釋其在生物通路(血管生成、細胞周期、DNA損傷反應、綜合癌癥通路、膠質母細胞瘤信號通路、TP53 通路)上的重要程度,以及富含癌癥驅動基因的良好表現。在本階段工作中,本文從復雜的蛋白質相互作用網絡中初步篩選出了腫瘤關鍵基因集,使得本文可以從該基礎上進一步地縮小規模,預測更為重要的腫瘤關鍵基因。
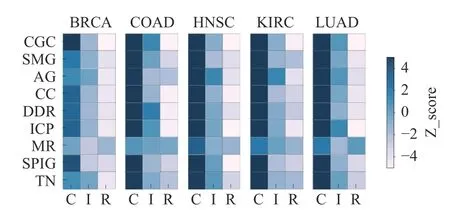
圖2 5 種癌癥Tumor 階段各類集合富集分析
對各類癌癥的多階段的關鍵基因集的作用情況進行統計,如圖3 所示,發現上述分類統計的腫瘤關鍵基因在整體中占比相近。為了進一步挖掘可預測的關鍵基因,本文利用顯著基因集(SMG,CGC)對這3 個部分進行富集占比分析,評估各個部分在腫瘤的突變過程中的作用效果,最終篩選出對腫瘤突變產生至關影響的部分。圖4 給出了各癌癥不同的集合在這兩個顯著基因集中的不同占比情況,可以發現:僅在Tumor 階段為關鍵基因的集合(Tumor-Normal)更具有研究意義,其存在于顯著基因集的占比在大多數情況下(BRCA、COAD、HNSC、KIRC、LUAD)都要優于另外兩個交集集合。相比顯著基因集在Normal 或者Tumor 中的占比,除了個別情況外(存在于癌癥的COAD、HNSC 集合中顯著基因集CGC 的占比),僅在Tumor 階段為關鍵基因的集合所存在的比例仍優于前者。考慮到單階段(Normal 或Tumor)的關鍵基因集,不能反應腫瘤在突變過程的變化,解釋不了其基因的突變以及對整個癌癥的影響趨勢,綜合Normal、Tumor 兩個階段選擇的關鍵基因集的研究要優于單階段的關鍵基因集。圖4 的COAD、HNSC 中,Normal 階段的關鍵基因集與僅在Tumor階段為關鍵基因的集合的比例相差在0.5%內,但這并不影響本文對結果(僅在Tumor 階段為關鍵基因的集合)的選擇。
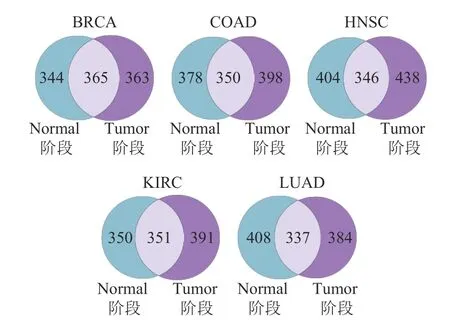
圖3 5 種癌癥類型Normal 階段和Tumor 階段關鍵基因集的韋恩圖
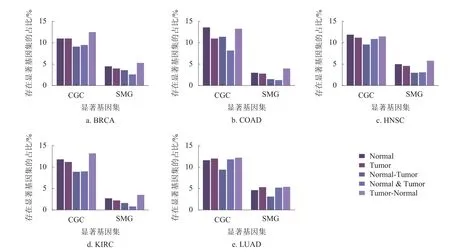
圖4 各癌癥各階段各部分的關鍵基因集與SMG、CGC 基因的占比情況
3.3 腫瘤關鍵基因分析
上述研究工作對腫瘤關鍵基因集進行了篩選,最終選擇了僅存在于Tumor 階段(Tumor-Normal)的腫瘤關鍵基因集,用于對驅動癌癥發展及突變的關鍵基因的預測。本文綜合了復雜網絡的度量指標,利用綜合中心性對最終的關鍵基因集按照分數進行降序排列,排名較高的基因更有可能驅動癌癥的發展,可作為潛在的靶點基因。接著利用SMG、CGC 顯著基因集,排除了已知被廣泛證實有驅動作用的一些關鍵基因。最后,對剩下的關鍵基因進行了分析,同時來驗證本文所預測的未知的驅動基因在癌癥發展變化過程中的顯著表現。
在乳腺癌(BRCA)的蛋白質相互作用網絡中,本文選取了綜合中心性排名靠前的SIRT7 基因作為驗證。圖5a 描述了SIRT7 的鄰居節點中SMG、CGC 基因的富集程度。為了排除一般基因(通常表現為低度)對結果的影響,實驗選取了排名在前40%的度較高的基因作為參考標準。其中圖5a 中的紅色標記為SIRT7 的鄰居中SMG 和CGC 的基因個數,紫色標記則為其他參考基因集與顯著基因集的交集基因分布情況。本文將交集個數大于等于觀察值的基因與總體的占比作為顯著性檢驗(p)的衡量標準。從結果可以看出,SIRT7 基因的鄰居節點相對于其他基因的鄰居節點,在兩個顯著基因集中都顯著富集(SMG 下p為4.73×10?3,CGC 下p為8.76×10?4)。實驗證明,SIRT7 對腫瘤網絡的控制有著重要作用,不僅從它作用性質的角度,而且可以通過它本身的突變帶動其周圍鄰居基因的突變,同時這些鄰居基因中富集了與癌癥相關的顯著基因。圖5b 展示了SIRT7 和它的鄰居基因的總體突變分布,其突變分布在T 檢驗中具有統計學意義(p=2.20×10?28),突變頻率明顯大于整體,在癌癥中具有較高的突變頻次。先前有實驗研究表明SIRT7 的表達可作為乳腺癌的預后生物標志物[28-29]。深圳大學健康科學中心的研究人員發現,SIRT7 與乳腺癌肺轉移相關,SIRT7 通過轉化因子?β 信號調節EMT,能夠抑制乳腺癌向肺部的轉移,提供了有效的乳腺癌轉移治療策略[28]。深圳大學附屬第一醫院的研究人員發現,SIRT7 可能是乳腺癌瘤體免疫浸潤相關的預后生物標志物[29]。
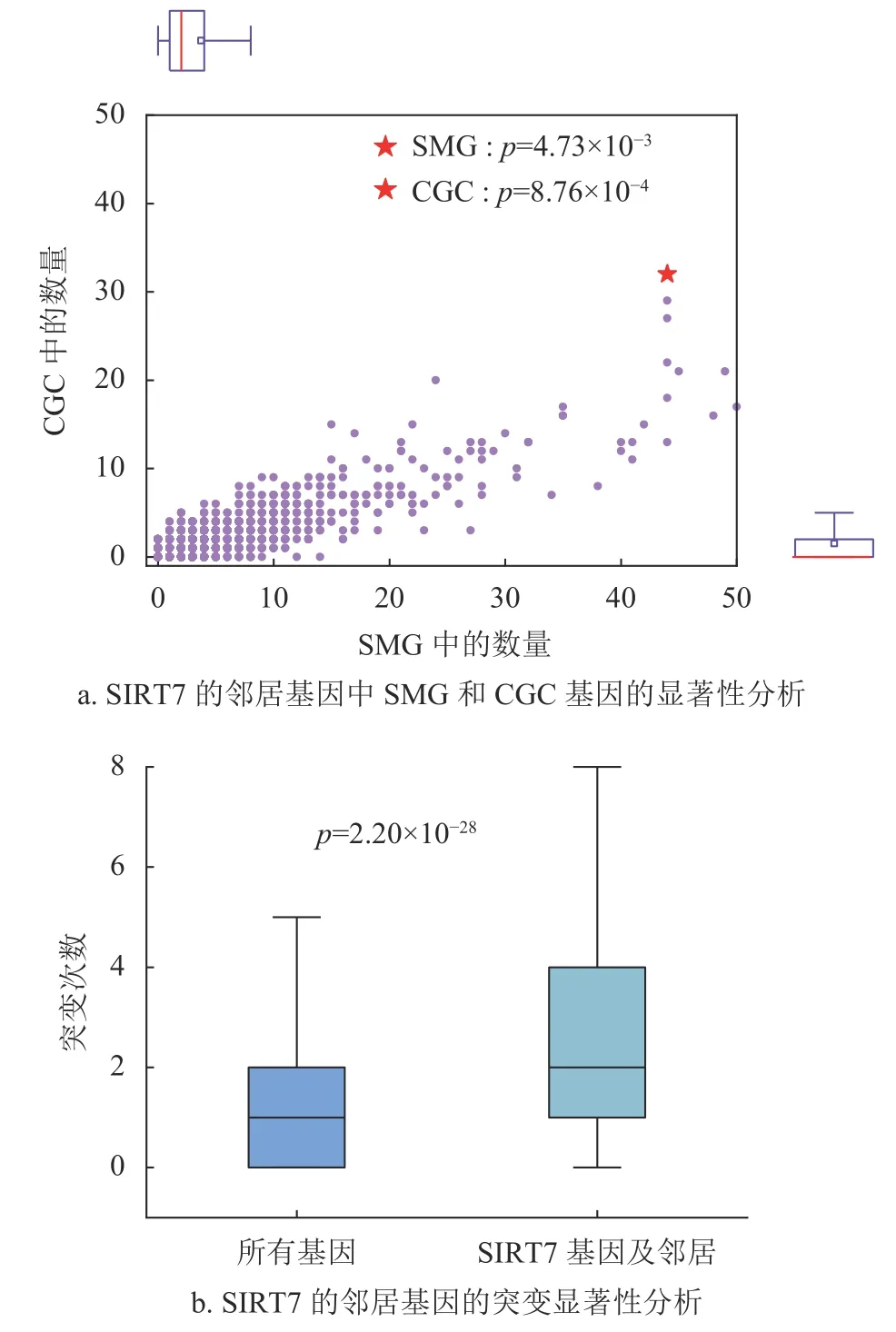
圖5 乳腺癌中的SIRT7 基因的重要性分析
在頭頸部鱗狀細胞癌(HNSC)的蛋白質相互作用網絡中,本文選擇了ETS1 基因作為驗證,圖6a描述了ETS1 基因的鄰居為SMG、CGC 基因的情況,結果表明ETS1 的鄰居顯著基因數要明顯多于所參考的基因集(SMG 下p為1.66×10?2,CGC 下p為4.56×10?2),這也驗證了基因的度中心性較高的基因更有可能與更多的顯著基因相鄰。ETS1 基因在癌癥中的表達變化,影響著與其相關的顯著基因,在發生癌變的過程中擔任著重要角色。圖6b顯示了ETS1 基因和它的鄰居相對于蛋白質相互作用網絡中所有基因及其鄰居的表現情況,結果表明ETS1 基因以及它的鄰居的突變分布要高于網絡整體水平(p=3.49×10?4),可作為致頭頸癌產生癌變的關鍵基因。已有研究表明ETS1可以作為治療頭頸癌的關鍵靶點[30-31]。美國紐約州立大學的研究人員發現ETS1 是頭頸部鱗狀細胞癌的生物標志物,它在頭頸部鱗狀細胞癌中的過度表達與預后不良相關,并且是關鍵上皮性向間質轉化(EMT)基因的主要調節因子,為腫瘤亞型特異性的靶向治療提供新途徑[30]。文獻[31]發現SRC-ETS1生存通路的上調與頭頸部鱗狀細胞癌HNSC 的細胞增殖、存活、遷移、侵襲和順鉑耐藥有關。
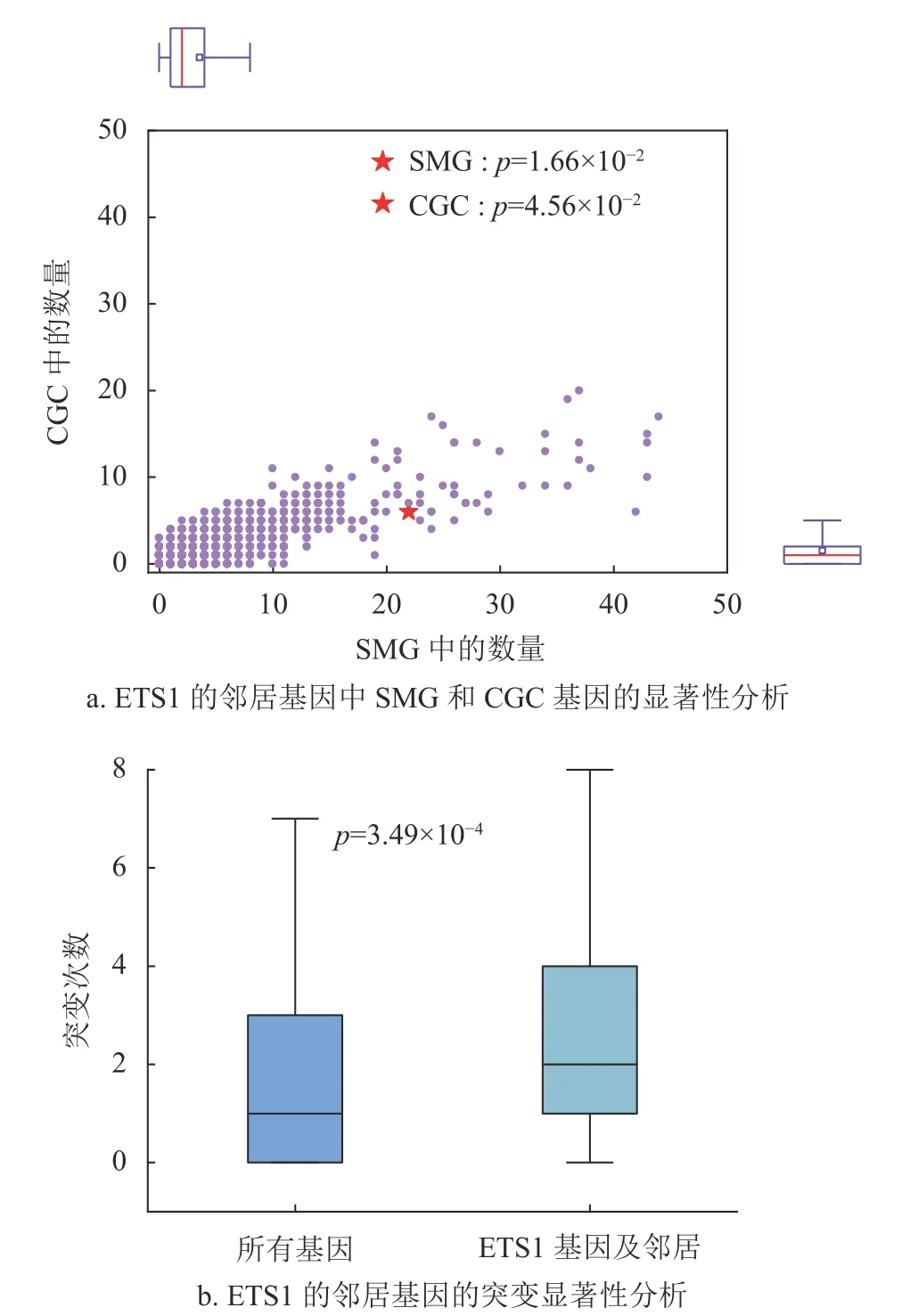
圖6 頭頸癌中的ETS1 基因的重要性分析
4 結束語
網絡控制理論能夠揭示預測蛋白質互作網絡的相互作用機制,能夠從復雜的生物網絡中識別出腫瘤關鍵基因,為癌癥的藥物設計及預后生物標志物預測提供了很好的借鑒。本文利用復雜網絡控制理論的方法對來自TCGA 數據庫中5 種癌癥網絡腫瘤基因進行了分類篩選,并結合腫瘤基因在兩個階段中的蛋白質網絡相互作用情況,進一步確認了腫瘤的關鍵基因集合。利用網絡的綜合中心性,除去已知被證實為顯著基因,對其他腫瘤關鍵基因集進行排序,得到潛在的腫瘤關鍵基因。通過驗證研究分析,在網絡中具有生物學統計意義的關鍵基因在腫瘤發展過程中的研究可能有著重要影響,它們能夠作為藥物靶點或者作為預后生物標志物,推動癌癥的治療及防控的研究。
本文利用復雜網絡控制理論和基因網絡分析,結合生物醫療數據對腫瘤的關鍵基因進行預測,為網絡控制在生物醫療方面的研究提供了良好的思路。但是本研究中只考慮了Normal 和Tumor 兩個階段的癌癥蛋白質相互作用網絡,如果進一步地結合Tumor 不同時期的數據細化腫瘤關鍵基因的識別,可以更好地進行腫瘤演化方面的分析,這也是未來工作中的研究方向。
猜你喜歡
中老年保健(2022年1期)2022-08-17 06:14:48
中學生數理化(高中版.高考理化)(2021年6期)2021-07-28 06:21:04
保健醫苑(2020年1期)2020-07-27 01:58:24
流行色(2020年9期)2020-07-16 08:08:32
人大建設(2019年9期)2019-12-27 09:06:30
當代水產(2019年1期)2019-05-16 02:42:14
NBA特刊(2014年7期)2014-04-29 00:44:03
傳記文學(2014年8期)2014-03-11 20:16:54
中國商人(2013年1期)2013-12-04 08:52:52
汽車與新動力(2012年5期)2012-03-25 10:09:44
